Lipid signaling
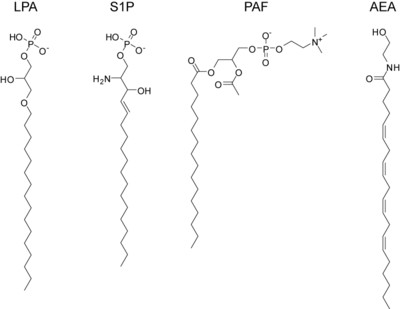
lysophosphatidic acid (LPA)
sphingosine-1-phosphate (S1P)
platelet activating factor (PAF)
anandamide or arachidonoyl ethanolamine (AEA)
Lipid signaling, broadly defined, refers to any biological signaling event involving a lipid messenger that binds a protein target, such as a receptor, kinase or phosphatase, which in turn mediate the effects of these lipids on specific cellular responses. Lipid signaling is thought to be qualitatively different from other classical signaling paradigms (such as monoamine neurotransmission) because lipids can freely diffuse through membranes (see osmosis.) One consequence of this is that lipid messengers cannot be stored in vesicles prior to release and so are often biosynthesized "on demand" at their intended site of action. As such, many lipid signaling molecules cannot circulate freely in solution but, rather, exist bound to special carrier proteins in serum.
Sphingolipid second messengers
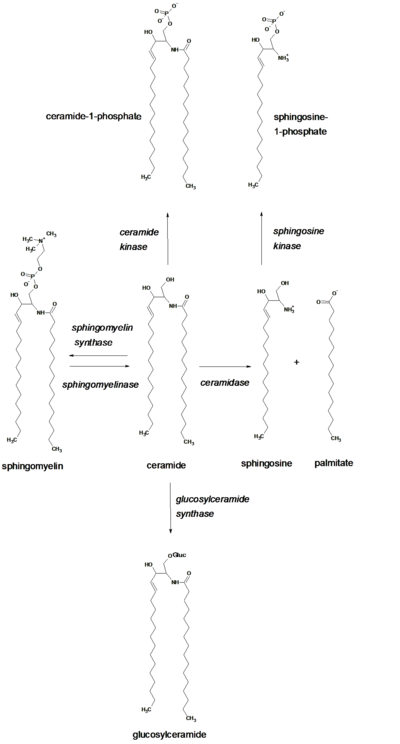
Ceramide
Ceramide (Cer) can be generated by the breakdown of sphingomyelin (SM) by sphingomyelinases (SMases), which are enzymes that hydrolyze the phosphocholine group from the sphingosine backbone. Alternatively, this sphingosine-derived lipid (sphingolipid) can be synthesized from scratch (de novo) by the enzymes serine palmitoyl transferase (SPT) and ceramide synthase in organelles such as the endoplasmic reticulum (ER) and possibly, in the mitochondria-associated membranes (MAMs) and the perinuclear membranes. Being located in the metabolic hub, ceramide leads to the formation of other sphingolipids, with the C1 hydroxyl (-OH) group as the major site of modification. A sugar can be attached to ceramide (glycosylation) through the action of the enzymes, glucosyl or galactosyl ceramide synthases.[1] Ceramide can also be broken down by enzymes called ceramidases, leading to the formation of sphingosine,[2][3] Moreover, a phosphate group can be attached to ceramide (phosphorylation) by the enzyme, ceramide kinase.[4] It is also possible to regenerate sphingomyelin from ceramide by accepting a phosphocholine headgroup from phosphatidylcholine (PC) by the action of an enzyme called sphingomyelin synthase.[5] The latter process results in the formation of diacylglycerol (DAG) from PC.
Ceramide contains two hydrophobic ("water-fearing") chains and a neutral headgroup. Consequently, it has limited solubility in water and is restricted within the organelle where it was formed. Also, because of its hydrophobic nature, ceramide readily flip-flops across membranes as supported by studies in membrane models and membranes from red blood cells (erythrocytes).[6] However, ceramide can possibly interact with other lipids to form bigger regions called microdomains which restrict its flip-flopping abilities. This could have immense effects on the signaling functions of ceramide because it is known that ceramide generated by acidic SMase enzymes in the outer leaflet of an organelle membrane may have different roles compared to ceramide that is formed in the inner leaflet by the action of neutral SMase enzymes.[7]
Ceramide mediates many cell-stress responses, including the regulation of programmed cell death (apoptosis) [8] and cell aging (senescence).[9] Numerous research works have focused interest on defining the direct protein targets of action of ceramide. These include enzymes called ceramide-activated Ser-Thr phosphatases (CAPPs), such as protein phosphatase 1 and 2A (PP1 and PP2A), which were found to interact with ceramide in studies done in a controlled environment outside of a living organism (in vitro).[10] On the other hand, studies in cells have shown that ceramide-inducing agents such as tumor necrosis factor-alpha α (TNFα) and palmitate induce the ceramide-dependent removal of a phosphate group (dephosphorylation) of the retinoblastoma gene product RB[11] and the enzymes, protein kinases B (AKT protein family) and C α (PKB and PKCα).[12] Moreover, there is also sufficient evidence which implicates ceramide to the activation of the kinase suppressor of Ras (KSR),[13] PKCζ,[14][15] and cathepsin D.[16] Interestingly, cathepsin D has been proposed as the main target for ceramide formed in organelles called lysosomes, making lysosomal acidic SMase enzymes one of the key players in the mitochondrial pathway of apoptosis. Ceramide was also shown to activate PKCζ, implicating it to the inhibition of AKT, regulation of the voltage difference between the interior and exterior of the cell (membrane potential) and signaling functions that favor apoptosis.[17] Chemotherapeutic agents such as daunorubicin and etoposide[18][19] enhance the de novo synthesis of ceramide in studies done on mammalian cells. The same results were found for certain inducers of apoptosis particularly stimulators of receptors in a class of lymphocytes (a type of white blood cell) called B-cells.[20] Regulation of the de novo synthesis of ceramide by palmitate may have a key role in diabetes and the metabolic syndrome. Experimental evidence shows that there is substantial increase of ceramide levels upon adding palmitate. Ceramide accumulation activates PP2A and the subsequent dephosphorylation and inactivation of AKT,[21] a crucial mediator in metabolic control and insulin signaling. This results in a substantial decrease in insulin responsiveness (i.e. to glucose) and in the death of insulin-producing cells in the pancreas called islets of Langerhans.[22] Inhibition of ceramide synthesis in mice via drug treatments or gene-knockout techniques prevented insulin resistance induced by fatty acids, glucocorticoids or obesity.[23]
An increase in in vitro activity of acid SMase has been observed after applying multiple stress stimuli such as ultraviolet (UV) and ionizing radiation, binding of death receptors and chemotherapeutic agents such as platinum, histone deacetylase inhibitors and paclitaxel.[24] In some studies, SMase activation results to its transport to the plasma membrane and the simultaneous formation of ceramide.[24]
Ceramide transfer protein (CERT) transports ceramide from ER to the Golgi for the synthesis of SM.[25] CERT is known to bind phosphatidylinositol phosphates, hinting its potential regulation via phosphorylation, a step of the ceramide metabolism that can be enzymatically regulated by protein kinases and phosphatases, and by inositol lipid metabolic pathways.[26] Up to date, there are at least 26 distinct enzymes with varied subcellular localizations, that act on ceramide as either a substrate or product. Regulation of ceramide levels can therefore be performed by one of these enzymes in distinct organelles by particular mechanisms at various times.[27]
Sphingosine
Sphingosine (Sph) is formed by the action of ceramidase (CDase) enzymes on ceramide in the lysosome. Sph can also be formed in the extracellular (outer leaflet) side of the plasma membrane by the action of neutral CDase enzyme. Sph then is either recycled back to ceramide or phosphorylated by one of the sphingosine kinase enzymes, SK1 and SK2.[28] The product sphingosine-1-phosphate (S1P) can be dephosphorylated in the ER to regenerate sphingosine by certain S1P phosphatase enzymes within cells, where the salvaged Sph is recycled to ceramide.[29] Sphingosine is a single-chain lipid (usually 18 carbons in length), rendering it to have sufficient solubility in water. This explains its ability to move between membranes and to flip-flop across a membrane. Estimates conducted at physiological pH show that approximately 70% of sphingosine remains in membranes while the remaining 30% is water-soluble.[30] Sph that is formed has sufficient solubility in the liquid found inside cells (cytosol). Thus, Sph may come out of the lysosome and move to the ER without the need for transport via proteins or membrane-enclosed sacs called vesicles. However, its positive charge favors partitioning in lysosomes. It is proposed that the role of SK1 located near or in the lysosome is to ‘trap’ Sph via phosphorylation.[31]
It is important to note that since sphingosine exerts surfactant activity, it is one of the sphingolipids found at lowest cellular levels.[31] The low levels of Sph and their increase in response to stimulation of cells, primarily by activation of ceramidase by growth-inducing proteins such as platelet-derived growth factor and insulin-like growth factor, is consistent with its function as a second messenger. It was found that immediate hydrolysis of only 3 to 10% of newly generated ceramide may double the levels of Sph.[31] Treatment of HL60 cells (a type of leukemia cell line) by a plant-derived organic compound called phorbol ester increased Sph levels threefold, whereby the cells differentiated into white blood cells called macrophages. Treatment of the same cells by exogenous Sph caused apoptosis. A specific protein kinase phosphorylates 14-3-3, otherwise known as sphingosine-dependent protein kinase 1 (SDK1), only in the presence of Sph.[32]
Sph is also known to interact with protein targets such as the protein kinase H homologue (PKH) and the yeast protein kinase (YPK). These targets in turn mediate the effects of Sph and its related sphingoid bases, with known roles in regulating the actin cytoskeleton, endocytosis, the cell cycle and apoptosis.[33] It is important to note however that the second messenger function of Sph is not yet established unambiguously.[34]
Sphingosine-1-Phosphate
Sphingosine-1-phosphate (S1P), like Sph, is composed of a single hydrophobic chain and has sufficient solubility to move between membranes. S1P is formed by phosphorylation of sphingosine by sphingosine kinase (SK). The phosphate group of the product can be detached (dephosphorylated) to regenerate sphingosine via S1P phosphatase enzymes or S1P can be broken down by S1P lyase enzymes to ethanolamine phosphate and hexadecenal.[35] Similar to Sph, its second messenger function is not yet clear.[34] However, there is substantial evidence that implicates S1P to cell survival, cell migration, and inflammation. Certain growth-inducing proteins such as platelet-derived growth factor (PDGF), insulin-like growth factor (IGF) and vascular endothelial growth factor (VEGF) promote the formation of SK enzymes, leading to increased levels of S1P. Other factors that induce SK include cellular communication molecules called cytokines, such as tumor necrosis factor α (TNFα) and interleukin-1 (IL-1), hypoxia or lack of oxygen supply in cells, oxidized low-density lipoproteins (oxLDL) and several immune complexes.[31]
S1P is probably formed at the inner leaflet of the plasma membrane in response to TNFα and other receptor activity-altering compounds called agonists.[36][37] S1P, being present in low nanomolar concentrations in the cell, has to interact with high-affinity receptors that are capable of sensing their low levels. So far, the only identified receptors for S1P are the high-affinity G protein-coupled receptors (GPCRs), also known as S1P receptors (S1PRs). S1P is required to reach the extracellular side (outer leaflet) of the plasma membrane to interact with S1PRs and launch typical GPCR signaling pathways.[38][39] However, the zwitterionic headgroup of S1P makes it unlikely to flip-flop spontaneously. To overcome this difficulty, the ATP-binding cassette (ABC) transporter C1 (ABCC1) serves as the "exit door" for S1P.[40] On the other hand, the cystic fibrosis transmembrane regulator (CFTR) serves as the means of entry for S1P into the cell.[41] In contrast to its low intracellular concentration, S1P is found in high nanomolar concentrations in serum where it is bound to albumin and lipoproteins.[42] Inside the cell, S1P can induce calcium release independent of the S1PRs—the mechanism of which remains unknown. To date, the intracellular molecular targets for S1P are still unidentified.[31]
The SK1-S1P pathway has been extensively studied in relation to cytokine action, with multiple functions connected to effects of TNFα and IL-1 favoring inflammation. Studies show that knockdown of key enzymes such as S1P lyase and S1P phosphatase increased prostaglandin production, parallel to increase of S1P levels.[37] This strongly suggests that S1P is the mediator of SK1 action and not subsequent compounds. Research done on endothelial and smooth muscle cells is consistent to the hypothesis that S1P has a crucial role in regulating endothelial cell growth, and movement.[43] Recent work on a sphingosine analogue, FTY270, demonstrates its ability to act as a potent compound that alters the activity of S1P receptors (agonist). FTY270 was further verified in clinical tests to have roles in immune modulation, such as that on multiple sclerosis.[44] This highlights the importance of S1P in the regulation of lymphocyte function and immunity. Most of the studies on S1P are used to further understand diseases such as cancer, arthritis and inflammation, diabetes, immune function and neurodegenerative disorders.[31]
Glucosylceramide
Glucosylceramides (GluCer) are the most widely distributed glycosphingolipids in cells serving as precursors for the formation of over 200 known glycosphingolipids. GluCer is formed by the glycosylation of ceramide in an organelle called Golgi via enzymes called glucosylceramide synthase (GCS) or by the breakdown of complex glycosphingolipids (GSLs) through the action of specific hydrolase enzymes. In turn, certain β-glucosidases hydrolyze these lipids to regenerate ceramide.[45][46] GluCer appears to be synthesized in the inner leaflet of the Golgi. Studies show that GluCer has to flip to the inside of the Golgi or transfer to the site of GSL synthesis to initiate the synthesis of complex GSLs. Transferring to the GSL synthesis site is done with the help of a transport protein known as four phosphate adaptor protein 2 (FAPP2) while the flipping to the inside of the Golgi is made possible by the ABC transporter P-glycoprotein, also known as the multi-drug resistance 1 transporter (MDR1).[47] GluCer is implicated in post-Golgi trafficking and drug resistance particularly to chemotherapeutic agents.[48][49] For instance, a study demonstrated a correlation between cellular drug resistance and modifications in GluCer metabolism.[50]
In addition to their role as building blocks of biological membranes, glycosphingolipids have long attracted attention because of their supposed involvement in cell growth, differentiation, and formation of tumors.[31] The production of GluCer from Cer was found to be important in the growth of neurons or brain cells.[51] On the other hand, pharmacological inhibition of GluCer synthase is being considered a technique to avoid insulin resistance.[52]
Ceramide-1-Phosphate
Ceramide-1-phosphate (C1P) is formed by the action of ceramide kinase (CK) enzymes on Cer. C1P carry ionic charge at neutral pH and contain two hydrophobic chains making it relatively insoluble in aqueous environment. Thus, C1P reside in the organelle where it was formed and is unlikely to spontaneously flip-flop across membrane bilayers.[31]
C1P activate phospholipase A2 and is found, along with CK, to be a mediator of arachidonic acid released in cells in response to a protein called interleukin-1β (IL-1β) and a lipid-soluble molecule that transports calcium ions (Ca2+) across the bilayer, also known as calcium ionophore.[53] C1P was also previously reported to encourage cell division (mitogenic) in fibroblasts, block apoptosis by inhibiting acid SMase in white blood cells within tissues (macrophages)[54] and increase intracellular free calcium concentrations in thyroid cells.[55] C1P also has known roles in vesicular trafficking, cell survival, phagocytosis ("cell eating") and macrophage degranulation.[56][57]
Phosphatidylinositol bisphosphate (PIP2) Lipid Agonist
PIP2 binds directly to ion channels and modulates their activity. PIP2 was shown to directly agonizes Inward rectifying potassium channels(Kir).[58] In this regard intact PIP2 signals as a bona fide neurotransmitter-like ligand. PIP2's interaction with many ion channels suggest that the intact form of PIP2 has an important signaling role independent of second messenger signaling.
Second messengers from phosphatidylinositol
Phosphatidylinositol bisphosphate (PIP2) Second Messenger Systems
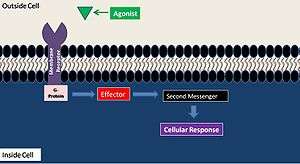
A general second messenger system mechanism can be broken down into four steps. First, the agonist activates a membrane-bound receptor. Second, the activated G-protein produces a primary effector. Third, the primary effect stimulates the second messenger synthesis. Fourth, the second messenger activates a certain cellular process.
The G-protein coupled receptors for the PIP2 messenger system produces two effectors, phospholipase C (PLC) and phosphoinositide 3-kinase (PI3K). PLC as an effector produces two different second messengers, inositol triphosphate (IP3) and Diacylglycerol (DAG).
IP3 is soluble and diffuses freely into the cytoplasm. As a second messenger, it is recognized by the inositol triphosphate receptor (IP3R), a Ca2+ channel in the endoplasmic reticulum (ER) membrane, which stores intracellular Ca2+. The binding of IP3 to IP3R releases Ca2+ from the ER into the normally Ca2+-poor cytoplasm, which then triggers various events of Ca2+ signaling. Specifically in blood vessels, the increase in Ca2+ concentration from IP3 releases nitric oxide, which then diffuses into the smooth muscle tissue and causes relaxation.[34]
DAG remains bound to the membrane by its fatty acid "tails" where it recruits and activates both conventional and novel members of the protein kinase C family. Thus, both IP3 and DAG contribute to activation of PKCs.[59][60]
Phosphoinositide 3-kinase (PI3K) as an effector phosphorylates phosphatidylinositol bisphosphate (PIP2) to produce phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 has been shown to activate protein kinase B, increase binding to extracellular proteins and ultimately enhance cell survival.[34]
Activators of G-protein coupled receptors
See main article on G-protein coupled receptors
Lysophosphatidic acid (LPA)
LPA is the result of phospholipase A2 action on phosphatidic acid. The SN-1 position can contain either an ester bond or an ether bond, with ether LPA being found at elevated levels in certain cancers. LPA binds the high-affinity G-protein coupled receptors LPA1, LPA2, and LPA3 (also known as EDG2, EDG4, and EDG7, respectively).
Sphingosine-1-phosphate (S1P)
S1P is present at high concentrations in plasma and secreted locally at elevated concentrations at sites of inflammation. It is formed by the regulated phosphorylation of sphingosine. It acts through five dedicated high-affinity G-protein coupled receptors, S1P1 - S1P5. Interestingly, targeted deletion of S1P1 results in lethality in mice and deletion of S1P2 results in seizures and deafness. Additionally, a mere 3- to 5-fold elevation in serum S1P concentrations induces sudden cardiac death by an S1P3-receptor specific mechanism.
Platelet activating factor (PAF)
PAF is a potent activator of platelet aggregation, inflammation, and anaphylaxis. It is similar to the ubiquitous membrane phospholipid phosphatidylcholine except that it contains an acetyl-group in the SN-2 position and the SN-1 position contains an ether-linkage. PAF signals through a dedicated G-protein coupled receptor, PAFR and is inactivated by PAF acetylhydrolase.
Endocannabinoids
The endogenous cannabinoids, or endocannabinoids, are endogenous lipids that activate cannabinoid receptors. The first such lipid to be isolated was anandamide which is the arachidonoyl amide of ethanolamine. Anandamide is formed via enzymatic release from N-arachidonoyl phosphatidylethanolamine by the N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD).[61] Anandamide activates both the CB1 receptor, found primarily in the central nervous system, and the CB2 receptor which is found primarily in lymphocytes and the periphery. It is found at very low levels (nM) in most tissues and is inactivated by the fatty acid amide hydrolase. Subsequently, another endocannabinoid was isolated, 2-arachidonoylglycerol, which is produced when phospholipase C releases diacylglycerol which is then converted to 2-AG by diacylglycerol lipase. 2-AG can also activate both cannabinoid receptors and is inactivated by monoacylglycerol lipase. It is present at approximately 100-times the concentration of anandamide in most tissues. Elevations in either of these lipids causes analgesia and anti-inflammation and tissue protection during states of ischemia, but the precise roles played by these various endocannabinoids are still not totally known and intensive research into their function, metabolism, and regulation is ongoing. One saturated lipid from this class, often called an endocannabinoid, but with no relevant affinity for the CB1 and CB 2 receptor is palmitoylethanolamide. This signaling lipid has great affinity for the GRP55 receptor and the PPAR alpha receptor. It has been identified as an anti-inflammatory compound already in 1957, and as an analgesic compound in 1975. It was Rita Levi-Montalcini. who first identified one of its biological mechanisms of action, the inhibition of activated mast cells. Palmitoylethanolamide is the only endocannabinoid available on the market for treatment, as a food supplement.
Prostaglandins
Prostaglandins are formed through oxidation of arachidonic acid by cyclooxygenases and other prostaglandin synthases. There are currently nine known G-protein coupled receptors (eicosanoid receptors) that largely mediate prostaglandin physiology (although some prostaglandins activate nuclear receptors, see below).
FAHFA
FAHFAs (fatty acid esters of hydroxy fatty acids) are formed in adipose tissue, improve glucose tolerance and also reduce adipose tissue inflammation. Palmitic acid esters of hydroxy-stearic acids (PAHSAs) are among the most bioactive members able to activate G-protein coupled receptors 120.[62] Docosahexaenoic acid ester of hydroxy-linoleic acid (DHAHLA) exert anti-inflammatory and pro-resolving properties.[63]
Retinol derivatives
Retinaldehyde is a retinol (vitamin A) derivative responsible for vision. It binds rhodopsin, a well-characterized GPCR that binds all-cis retinal in its inactive state. Upon photoisomerization by a photon the cis-retinal is converted to trans-retinal causing activation of rhodopsin which ultimately leads to depolarization of the neuron thereby enabling visual perception.
Activators of nuclear receptors
See the main article on nuclear receptors
Steroid Hormones
This large and diverse class of steroids are biosynthesized from isoprenoids and structurally resemble cholesterol. Mammalian steroid hormones can be grouped into five groups by the receptors to which they bind: glucocorticoids, mineralocorticoids, androgens, estrogens, and progestogens.
Retinoic acid
Retinol (vitamin A) can be metabolized to retinoic acid which activates nuclear receptors such as the RAR to control differentiation and proliferation of many types of cells during development.[64]
Prostaglandins
The majority of prostaglandin signaling occurs via GPCRs (see above) although certain prostaglandins activate nuclear receptors in the PPAR family. (See article eicosanoid receptors for more information).
See also
References
- ↑ Raas-Rothschild, A.; Pankova-Kholmyansky, I.; Kacher, Y.; Futerman, A. H. (2004). "Glycosphingolipidoses: beyond the enzymatic defect". Glycoconj. J. 21 (6): 295–304. doi:10.1023/B:GLYC.0000046272.38480.ef. PMID 15514478.
- ↑ Xu, R.; et al. (2006). "Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P". FASEB J. 20 (11): 1813–1825. doi:10.1096/fj.05-5689com. PMID 16940153.
- ↑ Galadari, S.; et al. (2006). "Identification of a novel amidase motif in neutral ceramidase". Biochem. J. 393 (Pt 3): 687–695. doi:10.1042/BJ20050682. PMC 1360721
 . PMID 16229686.
. PMID 16229686. - ↑ Wijesinghe DS, et al. (2005). "Substrate specificity of human ceramide kinase". J. Lipid Res. 46 (12): 2706–2716. doi:10.1194/jlr.M500313-JLR200. PMID 16170208.
- ↑ Tafesse, F. G.; Ternes, P.; Holthuis, J. C. (2006). "The multigenic sphingomyelin synthase family". J. Biol. Chem. 281 (40): 29421–29425. doi:10.1074/jbc.R600021200. PMID 16905542.
- ↑ Lopez-Montero, I.; et al. (2005). "Rapid transbilayer movement of ceramides in phospholipid vesicles and in human erythrocytes". J. Biol. Chem. 280 (27): 25811–25819. doi:10.1074/jbc.M412052200. PMID 15883154.
- ↑ Marchesini, N.; Hannun, Y. A. (2004). "Acid and neutral sphingomyelinases: roles and mechanisms of regulation". Biochem. Cell Biol. 82 (1): 27–44. doi:10.1139/o03-091. PMID 15052326.
- ↑ Obeid, L. M., Linardic, C. M., Karolak, L. A. & Hannun, Y. A. (1993) Programmed cell death induced by ceramide. Science. 259, 1769–1771 .
- ↑ Venable, M. E.; Lee, J. Y.; Smyth, M. J.; Bielawska, A.; Obeid, L. M. (1995). "Role of ceramide in cellular senescence". J. Biol. Chem. 270 (51): 30701–30708. doi:10.1074/jbc.270.51.30701. PMID 8530509.
- ↑ Chalfant, C. E.; Szulc, Z.; Roddy, P.; Bielawska, A.; Hannun, Y. A. (2004). "The structural requirements for ceramide activation of serine–threonine protein phosphatases". J. Lipid Res. 45 (3): 496–506. doi:10.1194/jlr.M300347-JLR200. PMID 14657198.
- ↑ Dbaibo, G.; et al. (1995). "Rb as a downstream target for a ceramide-dependent pathway of growth arrest". Proc. Natl. Acad. Sci. USA. 92 (5): 1347–1351. doi:10.1073/pnas.92.5.1347. PMC 42516. PMID 7877980.
- ↑ Lee, J. Y.; Hannun, Y. A.; Obeid, L. M. (1996). "Ceramide inactivates cellular protein kinase Cα". J. Biol. Chem. 271 (22): 13169–13174. doi:10.1074/jbc.271.22.13169. PMID 8662781.
- ↑ Zhang YH, et al. (1997). "Kinase suppressor of Ras is ceramide-activated protein kinase". Cell. 89 (1): 63–72. doi:10.1016/S0092-8674(00)80183-X. PMID 9094715.
- ↑ Mόller, G.; et al. (1995). "PKCζ is a molecular switch in signal transduction of TNF-α, bifunctionally regulated by ceramide and arachidonic acid". EMBO J. 14 (9): 1961–1969. PMC 398295. PMID 7744003.
- ↑ Bourbon, N. A.; Sandirasegarane, L.; Kester, M. (2002). "Ceramide-induced inhibition of Akt is mediated through protein kinase Cζ: implications for growth arrest". J. Biol. Chem. 277 (5): 3286–3292. doi:10.1074/jbc.M110541200. PMID 11723139.
- ↑ Heinrich, M.; et al. (2004). "Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation". Cell Death Differ. 11 (5): 550–563. doi:10.1038/sj.cdd.4401382. PMID 14739942.
- ↑ Wang, G.; et al. (2005). "Direct binding to ceramide activates protein kinase Cζ before the formation of a pro-apoptotic complex with PAR-4 in differentiating stem cells". J. Biol. Chem. 280 (28): 26415–26424. doi:10.1074/jbc.M501492200. PMID 15901738.
- ↑ Bose, R.; et al. (1995). "Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals". Cell. 82 (3): 405–414. doi:10.1016/0092-8674(95)90429-8. PMID 7634330.
- ↑ Perry DK, et al. (2000). "Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis". J. Biol. Chem. 275 (12): 9078–9084. doi:10.1074/jbc.275.12.9078. PMID 10722759.
- ↑ Kroesen BJ, et al. (2003). "BcR-induced apoptosis involves differential regulation of C16 and C24-ceramide formation and sphingolipid-dependent activation of the proteasome". J. Biol. Chem. 278 (17): 14723–14731. doi:10.1074/jbc.M210756200. PMID 12578840.
- ↑ Zhou, H. L.; Summers, S. K.; Birnbaum, M. J.; Pittman, R. N. (1998). "Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis". J. Biol. Chem. 273 (26): 16568–16575. doi:10.1074/jbc.273.26.16568. PMID 9632728.
- ↑ Unger, R. H. (2003). "Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome". Endocrinology. 144 (12): 5159–5165. doi:10.1210/en.2003-0870. PMID 12960011.
- ↑ Holland WL, et al. (2007). "Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance". Cell Metab. 5 (3): 167–179. doi:10.1016/j.cmet.2007.01.002. PMID 17339025.
- 1 2 Rotolo JA, et al. (2005). "Caspase-dependent and independent activation of acid sphingomyelinase signaling". J. Biol. Chem. 280 (28): 26425–26434. doi:10.1074/jbc.M414569200. PMID 15849201.
- ↑ Hanada, K.; et al. (2003). "Molecular machinery for non-vesicular trafficking of ceramide". Nature. 426 (6968): 803–809. doi:10.1038/nature02188. PMID 14685229.
- ↑ Fugmann, T.; et al. (2007). "Regulation of secretory transport by protein kinase D-mediated phosphorylation of the ceramide transfer protein". J. Cell Biol. 178 (1): 15–22. doi:10.1083/jcb.200612017. PMC 2064413. PMID 17591919.
- ↑ Hannun, Y.A.; Obeid, L.M. (2008). "Principles of bioactive lipid signalling: lessons from Sphingolipids". Nature Reviews Molecular Cell Biology. 9 (2): 139–150. doi:10.1038/nrm2329. PMID 18216770.
- ↑ Hait, N. C.; Oskeritzian, C. A.; Paugh, S. W.; Milstien, S.; Spiegel, S. (2006). "Sphingosine kinases, sphingosine 1 phosphate, apoptosis and diseases". Biochim. Biophys. Acta. 1758 (12): 2016–2026. doi:10.1016/j.bbamem.2006.08.007. PMID 16996023.
- ↑ Johnson KR, et al. (2003). "Role of human sphingosine-1-phosphate phosphatase 1 in the regulation of intra- and extracellular sphingosine-1-phosphate levels and cell viability". J. Biol. Chem. 278 (36): 34541–34547. doi:10.1074/jbc.M301741200. PMID 12815058.
- ↑ Khan WA, et al. (1991). "Use of d-erythro-sphingosine as a pharmacologic inhibitor of protein kinase C in human platelets". Biochem. J. 278: 387–392. PMC 1151354. PMID 1898331.
- 1 2 3 4 5 6 7 8 Hannun and Obeid (2008)
- ↑ Hamaguchi, A.; et al. (2003). "A sphingosine-dependent protein kinase that specifically phosphorylates 14-3-3 (SDK1) is identified as the kinase domain of PKC: a preliminary note. Biochemical and". Biophys. Res. Comm. 307 (3): 589–594. doi:10.1016/S0006-291X(03)01070-2.
- ↑ Smith, E. R.; Merrill, A. H.; Obeid, L. M.; Hannun, Y. A. (2000). "Effects of sphingosine and other sphingolipids on protein kinase C". Methods Enzymol. Methods in Enzymology. 312: 361–373. doi:10.1016/S0076-6879(00)12921-0. ISBN 9780121822132. PMID 11070884.
- 1 2 3 4 Prokazova, N.; et al. (2007). "Lipid second messengers and cell signaling in vascular wall". Biochemistry (Mosc.). 72 (8): 797–808. doi:10.1134/S0006297907080019.
- ↑ Bandhuvula, P.; Saba, J. D. (2007). "Sphingosine-1-phosphate lyase in immunity and cancer: silencing the siren". Trends Mol. Med. 13 (5): 210–217. doi:10.1016/j.molmed.2007.03.005. PMID 17416206.
- ↑ Xia, P.; et al. (1998). "Tumor necrosis factor-α induces adhesion molecule expression through the sphingosine kinase pathway". Proc. Natl. Acad. Sci. USA. 95: 14196–14201. doi:10.1073/pnas.95.24.14196.
- 1 2 Pettus BJ, et al. (2003). "The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-α". FASEB J. 17 (11): 1411–1421. doi:10.1096/fj.02-1038com. PMID 12890694.
- ↑ Hla, T.; Lee, M. J.; Ancellin, N.; Paik, J. H.; Kluk, M. J. (2001). "Lysophospholipids — receptor revelations". Science. 294 (5548): 1875–1878. doi:10.1126/science.1065323. PMID 11729304.
- ↑ Taha, T. A.; Argraves, K. M.; Obeid, L. M. "Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy". Biochim. Biophys. Acta. 1682: 48–55. doi:10.1016/j.bbalip.2004.01.006.
- ↑ Mitra, P.; et al. (2006). "Role of ABCC1 in export of sphingosine-1-phosphate from mast cells". Proc. Natl. Acad. Sci. USA. 103 (44): 16394–16399. doi:10.1073/pnas.0603734103. PMC 1637593. PMID 17050692.
- ↑ Boujaoude LC, et al. (2001). "Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: modulation of cellular activity of sphingosine 1-phosphate". J. Biol. Chem. 276 (38): 35258–35264. doi:10.1074/jbc.M105442200. PMID 11443135.
- ↑ Okajima, F. "Plasma lipoproteins behave as carriers of extracellular sphingosine 1-phosphate: is this an atherogenic mediator or an anti-atherogenic mediator?". Biochim. Biophys. Acta. 1582: 132–137. doi:10.1016/s1388-1981(02)00147-6.
- ↑ Peters, S. L.; Alewijnse, A. E. (2007). "Sphingosine-1-phosphate signaling in the cardiovascular system". Current Opinion in Pharmacology. 7 (2): 186–192. doi:10.1016/j.coph.2006.09.008. PMID 17280869.
- ↑ Gonsette, R. E. (2004). "New immunosuppressants with potential implication in multiple sclerosis". J. Neurol. Sci. 223 (1): 87–93. doi:10.1016/j.jns.2004.04.025. PMID 15261567.
- ↑ Hakomori, S. "Traveling for the glycosphingolipid path". Glycoconj. J. 17: 627–647.
- ↑ Ichikawa, S.; Hirabayashi, Y. "Glucosylceramide synthase and glycosphingolipid synthesis". Trends Cell Biol. 8: 198–202. doi:10.1016/s0962-8924(98)01249-5.
- ↑ D'Angelo, G.; et al. (2007). "Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide". Nature. 449 (7158): 62–67. doi:10.1038/nature06097. PMID 17687330.
- ↑ Radin, N. S., Shayman, J.A. & Inokuchi, J.-I. Metabolic effects of inhibiting glucosylceramide synthesis with PDMP and other substances. Adv. Lipid Res. 26, 183–211
- ↑ Gouaze-Andersson, V.; Cabot, M. C. "Glycosphingolipids and drug resistance". Biochim. Biophys. Acta. 1758: 2096–2103. doi:10.1016/j.bbamem.2006.08.012.
- ↑ Lavie, Y.; et al. (1996). "Accumulation of glucosylceramides in multidrug-resistant cancer cells". J. Biol. Chem. 271 (32): 19530–19536. doi:10.1074/jbc.271.32.19530. PMID 8702646.
- ↑ Schwarz, A.; Futerman, A. "Distinct roles for ceramide and glucosylceramide at different stages of neuronal growth". J. Neurosci. 17: 2929–2938.
- ↑ Aerts, J.; et al. (2007). "Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity". Diabetes. 56 (5): 1341–1349. doi:10.2337/db06-1619. PMID 17287460.
- ↑ Pettus BJ, et al. (2004). "Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2". J. Biol. Chem. 279 (12): 11320–11326. doi:10.1074/jbc.M309262200. PMID 14676210.
- ↑ Gomez-Munoz, A.; et al. (2004). "Ceramide-1-phosphate blocks apoptosis through inhibition of acid sphingomyelinase in macrophages". J. Lipid Res. 45 (1): 99–105. doi:10.1194/jlr.M300158-JLR200. PMID 14523050.
- ↑ Tornquist, K. (February 2003). "Ceramide-1-phosphate increases intracellular free calcium concentrations in thyroid FRTL-5 cells: evidence for an effect mediated by inositol-1,4,5-trisphosphate and intracellular sphingosine-1-phosphate". J. Biochem. 370 (Pt 1): 111–119. doi:10.1042/BJ20020970. PMC 1223145. PMID 12416995.
- ↑ Shayman, J.; et al. (2005). "Ceramide-1-phosphate, a mediator of phagocytosis". J. Biol. Chem. 280 (28): 26612–26621. doi:10.1074/jbc.M501359200. PMID 15899891.
- ↑ Gomez-Munoz, A.; et al. (2005). "Ceramide-1-phosphate promotes cell survival through activation of the phosphatidylinositol-3-kinase/protein kinase B pathway". FEBS Letters. 579 (17): 3744–3750. doi:10.1016/j.febslet.2005.05.067. PMID 15978590.
- ↑ Hansen, S. (2011). "Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2.". Nature. 477 (7365): 495–498. doi:10.1038/nature10370. PMC 3324908. PMID 21874019.
- ↑ Irvine, R. (1992). "Inositol lipids in cell signaling". Current Opinion in Cell Biology. 4 (2): 212–9. doi:10.1016/0955-0674(92)90035-B. PMID 1318060.
- ↑ Nishizuka, Y. (1995). "Protein kinase C and lipid signaling for sustained cellular responses". FASEB J. 9 (7): 484–496. PMID 7737456.
- ↑ Magotti P, Bauer I, Igarashi M, Babagoli M, Marotta R, Piomelli D, Garau G (Dec 2014). "Structure of Human N-Acylphosphatidylethanolamine-Hydrolyzing Phospholipase D: Regulation of Fatty Acid Ethanolamide Biosynthesis by Bile Acids". Structure 24 (3): 598–604. doi:10.1016/j.str.2014.12.018. PMID 25684574.
- ↑ Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, Dhaneshwar AS, Hammarstedt A, Smith U, McGraw TE, Saghatelian A, Kahn BB. "Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects" Cell. 2014 Oct 9;159(2):318-32. doi: 10.1016/j.cell.2014.09.035. PMID 25303528
- ↑ Kuda O, Brezinova M, Rombaldova M, Slavikova B, Posta M, Beier P, Janovska P, Veleba J, Kopecky J Jr, Kudova E, Pelikanova T, Kopecky J. "Docosahexaenoic acid-derived fatty acid esters of hydroxy fatty acids (FAHFAs) with anti-inflammatory properties" Diabetes. 2016 Jun 16. pii: db160385.PMID 27313314
- ↑ Duester, G (September 2008). "Retinoic acid synthesis and signaling during early organogenesis". Cell. 134 (6): 921–31. doi:10.1016/j.cell.2008.09.002. PMC 2632951. PMID 18805086.