Tandem mass spectrometry

Tandem mass spectrometry, also known as MS/MS or MS2, involves multiple steps of mass spectrometry selection, with some form of fragmentation occurring in between the stages.[1] In a tandem mass spectrometer, ions are formed in the ion source and separated by mass-to-charge ratio in the first stage of mass spectrometry (MS1). Ions of a particular mass-to-charge ratio (precursor ions) are selected and fragment ions (product ions) are created by collision-induced dissociation, ion-molecule reaction, photodissociation, or other process. The resulting ions are then separated and detected in a second stage of mass spectrometry (MS2).
Instrumentation
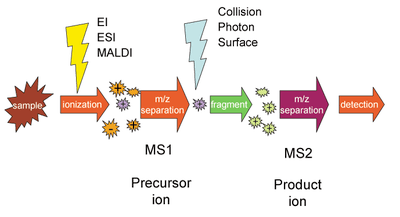
For tandem mass spectrometry in space, the different elements are often noted in shorthand. Multiple stages of mass analysis separation can be accomplished with individual mass spectrometer elements separated in space or using a single mass spectrometer with the MS steps separated in time.
Tandem in space
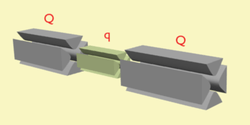
In tandem mass spectrometry in space, the separation elements are physically separated and distinct, although there is a physical connection between the elements to maintain high vacuum. These elements can be sectors, transmission quadrupole, or time-of-flight. When using multiple quadrupoles, they can act as both mass analyzers and collision chambers.
- Q – Quadrupole mass analyzer
- q – Radio frequency collision quadrupole
- TOF – Time-of-flight mass analyzer
- B – Magnetic sector
- E – Electric sector
The notation can be combined to indicate various hybrid instrument, for example
- QqQ – Triple quadrupole mass spectrometer
- QTOF – Quadrupole time-of-flight mass spectrometer (also QqTOF)
- BEBE – Four-sector (reverse geometry) mass spectrometer
Tandem in time

By doing tandem mass spectrometry in time, the separation is accomplished with ions trapped in the same place, with multiple separation steps taking place over time. A quadrupole ion trap or Fourier transform ion cyclotron resonance (FTICR) instrument can be used for such an analysis. Trapping instruments can perform multiple steps of analysis, which is sometimes referred to as MSn (MS to the n). Often the number of steps, n, is not indicated, but occasionally the value is specified; for example MS3 indicates three stages of separation. Tandem in time MS instruments do not use the modes described next, but typically collect all of the information from a precursor ion scan and a parent ion scan of the entire spectrum. Each instrumental configuration utilizes a unique mode of mass identification.
Tandem in space MS/MS modes
When tandem MS is performed with an in space design, the instrument must operate in one of a variety of modes. There are a number of different tandem MS/MS experimental setups and each mode has its own applications and provides different information. Tandem MS in space uses the coupling of two instrument components which measure the same mass spectrum range but with a controlled fractionation between them in space, while tandem MS in time involves the use of an ion trap.
There are four main scan experiments possible using MS/MS: precursor ion scan, product ion scan, neutral loss scan, and selected reaction monitoring.
For a precursor ion scan, the product ion is selected in the second mass analyzer, and the precursor masses are scanned in the first mass analyzer. Note that precursor ion[2] is synonymous with parent ion[3] and product ion[4] with daughter ion;[5] however the use of these anthropomorphic terms is discouraged.[6][7]
In a product ion scan, a precursor ion is selected in the first stage, allowed to fragment and then all resultant masses are scanned in the second mass analyzer and detected in the detector that is positioned after the second mass analyzer. This experiment is commonly performed to identify transitions used for quantification by tandem MS.
In a neutral loss scan, the first mass analyzer scans all the masses. The second mass analyzer also scans, but at a set offset from the first mass analyzer.[8] This offset corresponds to a neutral loss that is commonly observed for the class of compounds. In a constant-neutral-loss scan, all precursors that undergo the loss of a specified common neutral are monitored. To obtain this information, both mass analyzers are scanned simultaneously, but with a mass offset that correlates with the mass of the specified neutral. Similar to the precursor-ion scan, this technique is also useful in the selective identification of closely related class of compounds in a mixture.
In selected reaction monitoring, both mass analyzers are set to a selected mass. This mode is analogous to selected ion monitoring for MS experiments. A selective analysis mode, which can increase sensitivity.[9]
Fragmentation
Fragmentation of gas-phase ions is essential to tandem mass spectrometry and occurs between different stages of mass analysis. There are many methods used to fragment the ions and these can result in different types of fragmentation and thus different information about the structure and composition of the molecule.
In-source fragmentation
Often, the ionization process is sufficiently violent to leave the resulting ions with sufficient internal energy to fragment within the mass spectrometer. If the product ions persist in their non-equilibrium state for a moderate amount of time before auto-dissociation this process is called metastable fragmentation.[10][11] Nozzle-skimmer fragmentation refers to the purposeful induction of in-source fragmentation by increasing the nozzle-skimmer potential on usually electrospray based instruments. Although in-source fragmentation allows for fragmentation analysis, it is not technically tandem mass spectrometry unless metastable ions are mass analyzed or selected before auto-dissociation and a second stage of analysis is performed on the resulting fragments. In-source fragmentation is often used in addition to tandem mass spectrometry (with post-source fragmentation) to allow for two steps of fragmentation in a pseudo MS3-type of experiment.[12]
Collision-induced dissociation
Post-source fragmentation is most often what is being used in a tandem mass spectrometry experiment. Energy can also be added to the ions, which are usually already vibrationally excited, through post-source collisions with neutral atoms or molecules, the absorption of radiation, or the transfer or capture of an electron by a multiply charged ion. Collision-induced dissociation (CID), also called collisionally activated dissociation (CAD), involves the collision of an ion with a neutral atom or molecule in the gas phase and subsequent dissociation of the ion.[13][14] For example, consider

where the ion AB+ collides with the neutral species M and subsequently breaks apart. The details of this process are described by collision theory.
Higher-energy collisional dissociation (HCD) is a CID technique specific to orbitrap mass spectrometers in which fragmentation takes place external to the trap.[15]
Electron capture and transfer methods
The energy released when an electron is transferred to or captured by a multiply charged ion can induce fragmentation.
Electron capture dissociation
If an electron is added to a multiply charged positive ion, the Coulomb energy is liberated. Adding a free electron is called electron capture dissociation (ECD),[16] and is represented by
![{\displaystyle [{\ce {M}}+n{\ce {H}}]^{n+}+{\ce {e^{-}->}}\left[[{\ce {M}}+(n-1){\ce {H}}]^{(n-1)+}\right]^{*}{\ce {->fragments}}}](../I/m/80535d501352b7854827d81bd70a7811d203d639.svg)
for a multiply protonated molecule M.
Electron transfer dissociation
Adding an electron through an ion-ion reaction is called electron transfer dissociation (ETD).[17][18] Similar to electron-capture dissociation, ETD induces fragmentation of cations (e.g. peptides or proteins) by transferring electrons to them. It was invented by Donald F. Hunt, Joshua Coon, John E. P. Syka and Jarrod Marto at the University of Virginia.[19]
ETD does not use free electrons but employs radical anions (e.g. anthracene or azobenzene) for this purpose:
![{\displaystyle [{\ce {M}}+n{\ce {H}}]^{n+}+{\ce {A^{-}->}}\left[[{\ce {M}}+(n-1){\ce {H}}]^{(n-1)+}\right]^{*}+{\ce {A->fragments}}}](../I/m/a0f39c0e76fd3f9d3862b8b49cb144a43c1cd907.svg)
where A is the anion.[20]
ETD cleaves randomly along the peptide backbone (c and z ions) while side chains and modifications such as phosphorylation are left intact. The technique only works well for higher charge state ions (z>2), however relative to collision-induced dissociation (CID), ETD is advantageous for the fragmentation of longer peptides or even entire proteins. This makes the technique important for top-down proteomics. Much like ECD, ETD is effective for peptides with modifications such as phosphorylation.[21]
Electron-transfer and higher-energy collision dissociation (EThcD) is a combination ETD and HCD where the peptide precursor is initially subjected to an ion/ion reaction with fluoranthene anions in a linear ion trap, which generates c- and z-ions.[17][22] In the second step HCD all-ion fragmentation is applied to all ETD derived ions to generate b- and y- ions prior to final analysis in the orbitrap analyzer.[15] This method employs dual fragmentation to generate ion- and thus data-rich MS/MS spectra for peptide sequencing and PTM localization.[23]
Negative electron transfer dissociation
Fragmentation can also occur with a deprotonated species, in which an electron is transferred from the specie to an cationic reagent in a negative electron transfer dissociation (NETD):[24]
![{\displaystyle [{\ce {M}}-n{\ce {H}}]^{n-}+{\ce {A+->}}\left[[{\ce {M}}-n{\ce {H}}]^{(n+1)-}\right]^{*}+{\ce {A->fragments}}}](../I/m/383244602d9ce4607f68c0696b8c55fb10951a3f.svg)
Following this transfer event, the electron deficient anion undergoes internal rearrangement and fragments. NETD is the ion/ion analogue of electron-detachment dissociation (EDD).
NETD is compatible with fragmenting peptide and proteins along the backbone at the Cα-C bond. The resulting fragments are usually a•- and x-type product ions.
Electron-detachment dissociation
Electron-detachment dissociation (EDD) is a method for fragmenting anionic species in mass spectrometry.[25] It serves as a negative counter mode to electron capture dissociation. Negatively charged ions are activated by irradiation with electrons of moderate kinetic energy. The result is ejection of electrons from the parent ionic molecule, which causes dissociation via recombination.
Charge transfer dissociation
Reaction between positively charged peptides and cationic reagents,[26] also known as charge transfer dissociation (CTD),[27] has recently been demonstrated as an alternative high-energy fragmentation pathway for low-charge state (1+ or 2+) peptides. The proposed mechanism of CTD using helium cations as the reagent is:
![{\displaystyle {\ce {{[{M}+H]^{1}+}+He+->}}\left[{\ce {[{M}+H]^{2}+}}\right]^{*}+{\ce {He^{0}->fragments}}}](../I/m/a8ccd097f31299c546cd6e591efb6ed4f8556e2e.svg)
Initial reports are that CTD causes backbone Cα-C bond cleavage of peptides and provides a•- and x-type product ions.
Photodissociation
The energy required for dissociation can be added by photon absorption, resulting in ion photodissociation and represented by

where represents the photon absorbed by the ion. Ultraviolet lasers can be used, but can lead to excessive fragmentation of biomolecules.[28]

Infrared multiphoton dissociation
Infrared photons will heat the ions and cause dissociation if enough of them are absorbed. This process is called infrared multiphoton dissociation (IRMPD) and is often accomplished with a carbon dioxide laser and an ion trapping mass spectrometer such as a FTMS.[29]
Blackbody infrared radiative dissociation
Blackbody radiation can be used for photodissociation in a technique known as blackbody infrared radiative dissociation (BIRD).[30] In the BIRD method, the entire mass spectrometer vacuum chamber is heated to create infrared radiation. BIRD uses the light from black body radiation to thermally (vibrationally) excite the ions until a bond breaks.[30][31] This is similar to infrared multiphoton dissociation with the exception of the source of radiation.[14] This technique is most often used with Fourier transform ion cyclotron resonance mass spectrometers.
Surface induced dissociation
With surface-induced dissociation (SID), the fragmentation is a result of the collision of an ion with a surface under high vacuum.[32][33]
Quantitative proteomics
Quantitative proteomics is used to determine the relative or absolute amount of proteins in a sample.[34][35][36] Several quantitative proteomics methods are based on tandem mass spectrometry. MS/MS has become a benchmark procedure for the structural elucidation of complex biomolecules.[37]
Isobaric tag for relative and absolute quantitation
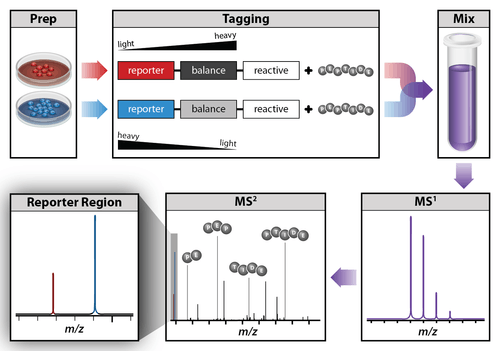
An isobaric tag for relative and absolute quantitation (iTRAQ) is a reagent for tandem mass spectrometry that is used to determine the amount of proteins from different sources in a single experiment.[38][39][40] It uses stable isotope labeled molecules that can form a covalent bond with the N-terminus and side chain amines of proteins. The iTRAQ reagents are used to label peptides from different samples that are pooled and analyzed by liquid chromatography and tandem mass spectrometry. The fragmentation of the attached tag generates a low molecular mass reporter ion that can be used to relatively quantify the peptides and the proteins from which they originated.
Tandem mass tag
A tandem mass tag (TMT) is an isobaric mass tag chemical label used for protein quantification and identification.[41] The tags contain four regions: mass reporter, cleavable linker, mass normalization, and protein reactive group.
Applications
Peptides
Tandem mass spectrometry can be used for protein sequencing.[42] When intact proteins are introduced to a mass analyzer, this is called "top-down proteomics" and when proteins are digested into smaller peptides and subsequently introduced into the mass spectrometer, this is called "bottom-up proteomics". Shotgun proteomics is a variant of bottom up proteomics in which proteins in a mixture are digested prior to separation and tandem mass spectrometry.
Tandem mass spectrometry can produce a peptide sequence tag that can be used to identify a peptide in a protein database.[43][44][45] A notation has been developed for indicating peptide fragments that arise from a tandem mass spectrum.[46] Peptide fragment ions are indicated by a, b, or c if the charge is retained on the N-terminus and by x, y or z if the charge is maintained on the C-terminus. The subscript indicates the number of amino acid residues in the fragment. Superscripts are sometimes used to indicate neutral losses in addition to the backbone fragmentation, * for loss of ammonia and ° for loss of water. Although peptide backbone cleavage is the most useful for sequencing and peptide identification other fragment ions may be observed under high energy dissociation conditions. These include the side chain loss ions d, v, w and ammonium ions[47][48] and additional sequence-specific fragment ions associated with particular amino acid residues.[49]
Oligosaccharides
Oligosaccharides may be sequenced using tandem mass spectrometry in a similar manner to peptide sequencing.[50] Fragmentation generally occurs on either side of the glycosidic bond (b, c, y and z ions) but also under more energetic conditions through the sugar ring structure in a cross-ring cleavage (x ions). Again trailing subscripts are used to indicate position of the cleavage along the chain. For cross ring cleavage ions the nature of the cross ring cleavage is indicated by preceding superscripts.[51][52]
Oligonucleotides
Tandem mass spectrometry has been applied to DNA and RNA sequencing.[53][54] A notation for gas-phase fragmentation of oligonucleotide ions has been proposed.[55]
Newborn screening
Newborn screening is the process of testing newborn babies for treatable genetic, endocrinologic, metabolic and hematologic diseases.[56][57] The development of tandem mass spectrometry screening in the early 1990s led to a large expansion of potentially detectable congenital metabolic diseases that affect blood levels of organic acids.[58]
See also
- Accelerator mass spectrometry
- Charge remote fragmentation
- Cross section (physics)
- Mass-analyzed ion kinetic energy spectrometry
- Unimolecular ion decomposition
References
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "tandem mass spectrometer".
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "precursor ion".
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "parent ion".
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "product ion".
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "daughter ion".
- ↑ Bursey, Maurice M. (1991). "Comment to readers: Style and the lack of it". Mass Spectrometry Reviews. 10: 1–2. doi:10.1002/mas.1280100102.
- ↑ Adams, J. (1992). "To the editor". Journal of the American Society for Mass Spectrometry. 3 (4): 473. doi:10.1016/1044-0305(92)87078-D.
- ↑ Louris JN, Wright LG, Cooks RG, Schoen AE (1985). "New scan modes accessed with a hybrid mass spectrometer". Analytical Chemistry. 57 (14): 2918–2924. doi:10.1021/ac00291a039.
- ↑ deHoffman, Edmond; Stroobant, Vincent (2003). Mass Spectrometry: Principles and Applications. Toronto: Wiley. p. 133. ISBN 0-471-48566-7.
- ↑ IUPAC gold book definition of metastable ion (in mass spectrometry)
- ↑ IUPAC gold book definition of transient (chemical) species
- ↑ Körner R, Wilm M, Morand K, Schubert M, Mann M (February 1996). "Nano electrospray combined with a quadrupole ion trap for the analysis of peptides and protein digests". Journal of the American Society for Mass Spectrometry. 7 (2): 150–6. doi:10.1016/1044-0305(95)00626-5. PMID 24203235.
- ↑ Wells JM, McLuckey SA (2005). "Collision-induced dissociation (CID) of peptides and proteins". Methods in Enzymology. 402: 148–85. doi:10.1016/S0076-6879(05)02005-7. PMID 16401509.
- 1 2 Sleno L, Volmer DA (October 2004). "Ion activation methods for tandem mass spectrometry". Journal of Mass Spectrometry. 39 (10): 1091–112. doi:10.1002/jms.703. PMID 15481084.
- 1 2 Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M (September 2007). "Higher-energy C-trap dissociation for peptide modification analysis". Nature Methods. 4 (9): 709–12. doi:10.1038/nmeth1060. PMID 17721543.
- ↑ Cooper HJ, Håkansson K, Marshall AG (2005). "The role of electron capture dissociation in biomolecular analysis". Mass Spectrometry Reviews. 24 (2): 201–22. doi:10.1002/mas.20014. PMID 15389856.
- 1 2 Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF (June 2004). "Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry". Proceedings of the National Academy of Sciences of the United States of America. 101 (26): 9528–33. Bibcode:2004PNAS..101.9528S. doi:10.1073/pnas.0402700101. PMC 470779
 . PMID 15210983.
. PMID 15210983. - ↑ Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JE, Shabanowitz J, Hunt DF (December 2006). "The utility of ETD mass spectrometry in proteomic analysis". Biochimica et Biophysica Acta. 1764 (12): 1811–22. doi:10.1016/j.bbapap.2006.10.003. PMC 1853258. PMID 17118725.
- ↑ US patent 7534622, Donald F. Hunt, Joshua J. Coon, John E.P. Syka, Jarrod A. Marto, "Electron transfer dissociation for biopolymer sequence mass spectrometric analysis", issued 2009-05-19
- ↑ McLuckey SA, Stephenson JL (1998). "Ion/ion chemistry of high-mass multiply charged ions". Mass Spectrometry Reviews. 17 (6): 369–407. doi:10.1002/(SICI)1098-2787(1998)17:6<369::AID-MAS1>3.0.CO;2-J. PMID 10360331.
- ↑ Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF (February 2007). "Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry". Proceedings of the National Academy of Sciences of the United States of America. 104 (7): 2193–8. Bibcode:2007PNAS..104.2193C. doi:10.1073/pnas.0607084104. PMC 1892997. PMID 17287358.
- ↑ Frese CK, Altelaar AF, van den Toorn H, Nolting D, Griep-Raming J, Heck AJ, Mohammed S (November 2012). "Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry". Analytical Chemistry. 84 (22): 9668–73. doi:10.1021/ac3025366. PMID 23106539.
- ↑ Frese CK, Zhou H, Taus T, Altelaar AF, Mechtler K, Heck AJ, Mohammed S (March 2013). "Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD)". Journal of Proteome Research. 12 (3): 1520–5. doi:10.1021/pr301130k. PMC 3588588. PMID 23347405.
- ↑ Coon JJ, Shabanowitz J, Hunt DF, Syka JE (June 2005). "Electron transfer dissociation of peptide anions". Journal of the American Society for Mass Spectrometry. 16 (6): 880–2. doi:10.1016/j.jasms.2005.01.015. PMID 15907703.
- ↑ Budnik BA, Haselmann KF, Zubarev RA (2001). "Electron detachment dissociation of peptide di-anions: an electron–hole recombination phenomenon". Chemical Physics Letters. 342 (3-4): 299–302. Bibcode:2001CPL...342..299B. doi:10.1016/S0009-2614(01)00501-2.
- ↑ Chingin K, Makarov A, Denisov E, Rebrov O, Zubarev RA (January 2014). "Fragmentation of positively-charged biological ions activated with a beam of high-energy cations". Analytical Chemistry. 86 (1): 372–9. doi:10.1021/ac403193k.
- ↑ Hoffmann WD, Jackson GP (November 2014). "Charge transfer dissociation (CTD) mass spectrometry of peptide cations using kiloelectronvolt helium cations". Journal of the American Society for Mass Spectrometry. 25 (11): 1939–43. Bibcode:2014JASMS.tmp..208H. doi:10.1007/s13361-014-0989-6.
- ↑ Morgan JW, Hettick JM, Russell DH (2005). "Peptide sequencing by MALDI 193-nm photodissociation TOF MS". Methods in Enzymology. 402: 186–209. doi:10.1016/S0076-6879(05)02006-9. PMID 16401510.
- ↑ Little DP, Speir JP, Senko MW, O'Connor PB, McLafferty FW (September 1994). "Infrared multiphoton dissociation of large multiply charged ions for biomolecule sequencing". Analytical Chemistry. 66 (18): 2809–15. doi:10.1021/ac00090a004. PMID 7526742.
- 1 2 Schnier PD, Price WD, Jockusch RA, Williams ER (July 1996). "Blackbody infrared radiative dissociation of bradykinin and its analogues: energetics, dynamics, and evidence for salt-bridge structures in the gas phase". Journal of the American Chemical Society. 118 (30): 7178–89. doi:10.1021/ja9609157. PMC 1393282. PMID 16525512.
- ↑ Dunbar RC (2004). "BIRD (blackbody infrared radiative dissociation): evolution, principles, and applications". Mass Spectrometry Reviews. 23 (2): 127–58. doi:10.1002/mas.10074. PMID 14732935.
- ↑ Grill V, Shen J, Evans C, Cooks RG (2001). "Collisions of ions with surfaces at chemically relevant energies: Instrumentation and phenomena". Review of Scientific Instruments. 72 (8): 3149. Bibcode:2001RScI...72.3149G. doi:10.1063/1.1382641.
- ↑ Mabud, M. (1985). "Surface-induced dissociation of molecular ions". International Journal of Mass Spectrometry and Ion Processes. 67 (3): 285–294. Bibcode:1985IJMSI..67..285M. doi:10.1016/0168-1176(85)83024-X.
- ↑ Ong SE, Mann M (October 2005). "Mass spectrometry-based proteomics turns quantitative". Nature Chemical Biology. 1 (5): 252–62. doi:10.1038/nchembio736. PMID 16408053.
- ↑ Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B (October 2007). "Quantitative mass spectrometry in proteomics: a critical review". Analytical and Bioanalytical Chemistry. 389 (4): 1017–31. doi:10.1007/s00216-007-1486-6. PMID 17668192.
- ↑ Nikolov M, Schmidt C, Urlaub H (2012). "Quantitative mass spectrometry-based proteomics: an overview". Methods in Molecular Biology. 893: 85–100. doi:10.1007/978-1-61779-885-6_7. PMID 22665296.
- ↑ Maher S, Jjunju FP, Taylor S (2015). "100 years of mass spectrometry: Perspectives and future trends". Rev. Mod. Phys. 87 (1): 113–135. Bibcode:2015RvMP...87..113M. doi:10.1103/RevModPhys.87.113.
- ↑ Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ (December 2004). "Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents". Molecular & Cellular Proteomics. 3 (12): 1154–69. doi:10.1074/mcp.M400129-MCP200. PMID 15385600.
- ↑ Zieske LR (2006). "A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies". Journal of Experimental Botany. 57 (7): 1501–8. doi:10.1093/jxb/erj168. PMID 16574745.
- ↑ Gafken PR, Lampe PD (2006). "Methodologies for characterizing phosphoproteins by mass spectrometry". Cell Communication & Adhesion. 13 (5-6): 249–62. doi:10.1080/15419060601077917. PMC 2185548. PMID 17162667.
- ↑ Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C (April 2003). "Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS". Analytical Chemistry. 75 (8): 1895–904. doi:10.1021/ac0262560. PMID 12713048.
- ↑ Angel TE, Aryal UK, Hengel SM, Baker ES, Kelly RT, Robinson EW, Smith RD (May 2012). "Mass spectrometry-based proteomics: existing capabilities and future directions". Chemical Society Reviews. 41 (10): 3912–28. doi:10.1039/c2cs15331a. PMC 3375054. PMID 22498958.
- ↑ Hardouin J (2007). "Protein sequence information by matrix-assisted laser desorption/ionization in-source decay mass spectrometry". Mass Spectrometry Reviews. 26 (5): 672–82. doi:10.1002/mas.20142. PMID 17492750.
- ↑ Shadforth I, Crowther D, Bessant C (November 2005). "Protein and peptide identification algorithms using MS for use in high-throughput, automated pipelines". Proteomics. 5 (16): 4082–95. doi:10.1002/pmic.200402091. PMID 16196103.
- ↑ Mørtz E, O'Connor PB, Roepstorff P, Kelleher NL, Wood TD, McLafferty FW, Mann M (August 1996). "Sequence tag identification of intact proteins by matching tanden mass spectral data against sequence data bases". Proceedings of the National Academy of Sciences of the United States of America. 93 (16): 8264–7. Bibcode:1996PNAS...93.8264M. doi:10.1073/pnas.93.16.8264. PMC 38658. PMID 8710858.
- ↑ Roepstorff P, Fohlman J (November 1984). "Proposal for a common nomenclature for sequence ions in mass spectra of peptides". Biomedical Mass Spectrometry. 11 (11): 601. doi:10.1002/bms.1200111109. PMID 6525415.
- ↑ Johnson RS, Martin SA, Biemann K (December 1988). "Collision-induced fragmentation of (M + H)+ ions of peptides. Side chain specific sequence ions". International Journal of Mass Spectrometry and Ion Processes. 86: 137–154. doi:10.1016/0168-1176(88)80060-0.
- ↑ Falick AM, Hines WM, Medzihradszky KF, Baldwin MA, Gibson BW (November 1993). "Low-mass ions produced from peptides by high-energy collision-induced dissociation in tandem mass spectrometry". Journal of the American Society for Mass Spectrometry. 4 (11): 882–93. doi:10.1016/1044-0305(93)87006-X. PMID 24227532.
- ↑ Downard KM, Biemann K (January 1995). "Methionine specific sequence ions formed by the dissociation of protonated peptides at high collision energies". Journal of Mass Spectrometry. 30 (1): 25–32. doi:10.1002/jms.1190300106.
- ↑ Zaia J (2004). "Mass spectrometry of oligosaccharides". Mass Spectrometry Reviews. 23 (3): 161–227. doi:10.1002/mas.10073. PMID 14966796.
- ↑ Bruno Domon; Catherine E Costello (1988). "A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates". Glycoconj. J. 5 (4): 397–409. doi:10.1007/BF01049915.
- ↑ Spina E, Cozzolino R, Ryan E, Garozzo D (August 2000). "Sequencing of oligosaccharides by collision-induced dissociation matrix-assisted laser desorption/ionization mass spectrometry". Journal of Mass Spectrometry. 35 (8): 1042–8. doi:10.1002/1096-9888(200008)35:8<1042::AID-JMS33>3.0.CO;2-Y. PMID 10973004.
- ↑ Banoub JH, Newton RP, Esmans E, Ewing DF, Mackenzie G (May 2005). "Recent developments in mass spectrometry for the characterization of nucleosides, nucleotides, oligonucleotides, and nucleic acids". Chemical Reviews. 105 (5): 1869–915. doi:10.1021/cr030040w. PMID 15884792.
- ↑ Thomas B, Akoulitchev AV (March 2006). "Mass spectrometry of RNA". Trends in Biochemical Sciences. 31 (3): 173–81. doi:10.1016/j.tibs.2006.01.004. PMID 16483781.
- ↑ Wu J, McLuckey SA (2004). "Gas-phase fragmentation of oligonucleotide ions". International Journal of Mass Spectrometry. 237 (2–3): 197–241. Bibcode:2004IJMSp.237..197W. doi:10.1016/j.ijms.2004.06.014.
- ↑ Tarini BA (August 2007). "The current revolution in newborn screening: new technology, old controversies". Archives of Pediatrics & Adolescent Medicine. 161 (8): 767–72. doi:10.1001/archpedi.161.8.767. PMID 17679658.
- ↑ Kayton A (2007). "Newborn screening: a literature review". Neonatal Network. 26 (2): 85–95. doi:10.1891/0730-0832.26.2.85. PMID 17402600.
- ↑ Chace DH, Kalas TA, Naylor EW (November 2003). "Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns". Clinical Chemistry. 49 (11): 1797–817. doi:10.1373/clinchem.2003.022178. PMID 14578311.
Bibliography
- McLuckey, Scott A.; Busch, Kenneth L.; Glish, Gary L. (1988). Mass spectrometry/mass spectrometry: techniques and applications of tandem mass spectrometry. New York, N.Y: VCH Publishers. ISBN 0-89573-275-0.
- McLuckey, Scott A.; Glish, Gary L. Mass Spectrometry/Mass Spectrometry: Techniques and Applications of Tandem. Chichester: John Wiley & Sons. ISBN 0-471-18699-6.
- McLafferty, Fred W. (1983). Tandem mass spectrometry. New York: Wiley. ISBN 0-471-86597-4.
- Sherman, Nicholas E.; Kinter, Michael (2000). Protein sequencing and identification using tandem mass spectrometry. New York: John Wiley. ISBN 0-471-32249-0.
External links
| Ion source | |
|---|---|
| Mass analyzer | |
| Detector | |
| MS combination | |
| Fragmentation | |
| |