SuperPose
| Content | |
|---|---|
| Description | For protein structure superposition |
| Contact | |
| Research center | University of Alberta |
| Laboratory | Dr. David Wishart |
| Primary citation | [1] |
| Release date | 2004 |
| Access | |
| Data format | Data input: PDB files of two structures to be superimposed. Data output: Sequence alignments, structure alignments, PDB (Protein Data Bank) coordinates and RMSD statistics, as well as difference distance plots and images (both static and interactive) of the superimposed molecules. |
| Website | http://wishart.biology.ualberta.ca/SuperPose/ |
SuperPose is a freely available web server designed to perform both pairwise and multiple protein structure superpositions.[1] The “Structural superposition” term refers to the rotations and translations performed on one structure to make it match or align with another structure or structures. Structural superposition can be quantified either in terms of similarity or difference measures. The optimal superposition is the one in which the similarity measure is maximized (the former case) or the difference measure (the later case) is minimized. The “SuperPose” web server uses “RMSD” or Root-Mean-Square Deviation as a difference measure to find the optimal pairwise or multiple protein structure superposition. After an initial sequence and secondary structure (in case of low sequence identity) alignment, SuperPose generates a Difference Distance (DD) matrix [2] from the equivalent C-alpha atoms of two molecules. The sequence/structure alignment and DD matrix analysis information is then fed into a modified quaternion eigenvalue algorithm [3] to rapidly perform the structural superposition and calculate the RMSD between aligned regions of two macromolecules.
Input/Output
The SuperPose web server requires PDB formatted files (of two or more protein structures to be superimposed) or their PDB accession numbers as input. SuperPose can handle both X-ray and NMR structures. NMR structures often consist of 20-30 near-identical structures that need to be superimposed on each other to create a multistructure “blurogram”. For a superposition of two or more structures, SuperPose generates sequence alignments, structure alignments, PDB (Protein Data Bank) coordinates and RMSD statistics, as well as difference distance plots and images (both static and interactive) of the superimposed molecules.[1] All superimposed structure images can be reformatted as wireframe or ribbon, colour or greyscale, stereo or mono using the Output Options menu. The background colour of the images can also be toggled from black to white.
General Scope
SuperPose is very flexible and is able to superimpose structures that have substantial differences in sequence, size or shape. As a result, it can handle a much larger variety of superposition queries and situations than most other programs or servers. In particular, SuperPose can handle superpositions with: (i) identical sequences but slightly different structures; (ii) identical sequences but profoundly different structures (e.g. open and closed forms of calmodulin); (iii) modestly dissimilar sequences, lengths and structures; (iv) different sequence lengths but similar structures or sequences; and (v) largely different sequences but largely similar structures. Superpose is able to calculate both pairwise and multiple structure superpositions. It can also generate average and pairwise RMSD values for alpha carbons, backbone atoms, heavy atoms and all atoms. In case of identical sequence comparison, SuperPose generates “per-residue” RMSD tables and plots that allow users to identify, assess and view individual residue shifts or positional displacements. The SuperPose web server is freely accessible at http://wishart.biology.ualberta.ca/SuperPose
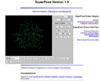 (a) |
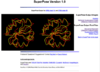 (b) |
 (c) |
 (d) |
 (e) |
Figure: Screenshot images of SuperPose server showing different kinds of graphical and textual outputs available. (a) A WebMol viewer, (b) a MolScript image, (c) a pairwise alignment, (d) a difference distance matrix and (e) the RMSD output for a pairwise superposition of 2TRX_A and 3TRX_A (Eschericia coli thioredoxin and human thioredoxin) are shown. Note that the sequence identity between two proteins is 29%.
See also
References
- 1 2 3 Maiti, Rajarshi; Gary H. Van Domselaar; Haiyan Zhang; David S. Wishart (July 2004). "SuperPose: a simple server for sophisticated structural superposition". Nucleic Acids Res. 32 (Web Server issue): W590–594. doi:10.1093/nar/gkh477. PMC 441615
 . PMID 15215457.
. PMID 15215457. - ↑ Richards, F.M.; Kundrot, C.E. (1988). "Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure". Proteins. 3 (2): 71–84. doi:10.1002/prot.340030202. PMID 3399495.
- ↑ Kearsley, S.K. (1990). "An algorithm for the simultaneous superposition of a structural series". J. Comput. Chem. 11 (10): 1187–1192. doi:10.1002/jcc.540111011.