Retrometabolic drug design
In the field of drug discovery, retrometabolic drug design is a strategy for the design of safer drugs using either a soft drug or targeted drug delivery approaches. The phrase retrometabolic drug design was coined by Nicholas Bodor.[1] The method is analogous to retrosynthetic analysis where the synthesis of a target molecule is planned backwards. In retrometabolic drug design, metabolic reaction information of drugs is used to design parent drugs whose metabolism and distribution can be controlled to target and eliminate the drug to increase efficacy and minimize undesirable side effects. The new drugs thus designed achieve selective organ and/or therapeutic site drug targeting and produce safe therapeutic agents and safe environmental chemicals. These approaches represent systematic methodologies that thoroughly integrate structure-activity (SAR) and structure-metabolism (SMR) relationships and are aimed at designing safe, locally active compounds with improved therapeutic index (ratio of benefit vs. side effect).[2][3][4][5][6]
Classification
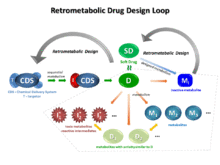
The concept of retrometabolic drug design encompasses two distinct approaches. One approach is the design of soft drugs (SDs),[4][7][8][9][10][11][12][13][14][15] new, active therapeutic agents, often isosteric or isolelectronic analogs of a lead compound, with a chemical structure specifically designed to allow predictable metabolism into inactive metabolites after exerting their desired therapeutic effect(s). The other approach is the design of chemical delivery systems (CDSs).[4][16][17][18][19][20][21][22][23] CDSs are biologically inert molecules intended to enhance drug delivery to a particular organ or site and requiring several conversion steps before releasing the active drug.
Although both retrometabolic design approaches involve chemical modifications of the molecular structure and both require enzymatic reactions to fulfill drug targeting, the principles of SD and CDS design are distinctly different. While CDSs are inactive as administered and sequential enzymatic reactions provide the differential distribution and ultimately release the active drug, SDs are active as administered and are designed to be easily metabolized into inactive species. Assuming an ideal situation, with a CDS the drug is present at the site and nowhere else in the body because enzymatic processes destroy the drug at those sites. Whereas, CDSs are designed to achieve drug targeting at a selected organ or site, SDs are designed to afford a differential distribution that can be regarded as reverse targeting.
Soft drugs
Since its introduction by Nicholas Bodor in the late 1970s, the soft drug concept generated considerable research both in academic and in industrial settings and there are several rationally designed soft drugs that have either already reached the market, such as
- esmolol (Breviblock)
- landiolol (Onoact)
- remifentanil (Ultiva)
- loteprednol etabonate (Lotemax, Alrex, Zylet)
- clevidipine (Cleviprex)
or are in late-stage development (remimazolam, budiodarone, celivarone, AZD3043, tecafarin).[24] There are also compounds that can be considered as soft chemicals (e.g., malathion) or soft drugs (e.g., articaine, methylphenidate)even though they were not developed as such.[24]
Chemical delivery systems
Since their introduction in the early 1980s, CDSs have also generated considerable research work, especially for brain and eye targeting of various therapeutic agents, including those that cannot cross the blood-brain barrier or the blood-retinal barrier on their own. Within this approach, three major general CDS classes have been identified:
- Enzymatic physicochemical-based (e.g., brain-targeting) CDSs: exploit site-specific traffic properties by sequential metabolic conversions that result in considerably altered properties
- Site-specific enzyme-activated (e.g., eye-targeting) CDSs: exploit specific enzymes found primarily, exclusively, or at higher activity at the site of action
- Receptor-based transient anchor-type (e.g., lung-targeting) CDSs: provide enhanced selectivity and activity through transient, reversible binding at the receptor
This concept has been extended to many drugs and peptides, its importance illustrated by the fact that its first applications and uses were published in Science[25][26][27] in 1975, 1981 and 1983. Its extension to the targeted brain-delivery of neuropeptides was included by the Harvard Health Letter[28] as one of the top 10 medical advances of 1992. Several compounds have reached advanced clinical development phase, such as
In the first example above, brain-targeted CDSs employ a sequential metabolic conversion of a redox-based targetor moiety, which is closely related to the ubiquitous NAD(P)H ⇌ NAD(P)+ coenzyme system, to exploit the unique properties of the blood-brain barrier (BBB). After enzymatic oxidation of the NADH type drug conjugate to its corresponding NAD+- drug, the still inactive precursor, "locks-in" behind the BBB to provide targeted and sustained CNS-delivery of the compound of interest.
The second example involves eye-specific delivery of betaxoxime, the oxime derivative of betaxolol. The administered, inactive β-amino-ketoxime is converted to the corresponding ketone via oxime hydrolase, an enzyme recently identified with preferential activity in the eye, and then stereospecifically reduced to its alcohol form. IOP-lowering activity is demonstrated without producing the active β-blockers systemically, making them void of any cardiovascular activity, a major drawback of classical antiglaucoma agents. Because of the advantages provided by this unique eye-targeting profile, oxime-based eye-targeting CDSs could replace the β-blockers currently used for ophthalmic applications.
History and significance
These retrometabolic design strategies were introduced by Nicholas Bodor, one of the first and most prominent advocates for the early integration of metabolism, pharmacokinetic and general physicochemical considerations in the drug design process.[31][32][33] These drug design concepts recognize the importance of design-controlled metabolism and directly focus not on the increase of activity alone but on the increase of the activity/toxicity ratio (therapeutic index) in order to deliver the maximum benefit while also reducing or eliminating unwanted side effects. The importance of this field is reviewed in a book dedicated to the subject (Bodor, N.; Buchwald, P.; Retrometabolic Drug Design and Targeting, 1st ed., Wiley & Sons, 2012), as well as by a full chapter of Burger's Medicinal Chemistry and Drug Design, 7th ed. (2010) with close to 150 chemical structures and more than 450 references.[34] At the time of its introduction, the idea of designed-in metabolism represented a significant novelty and was against mainstream thinking then in place that instead focused on minimizing or entirely eliminating drug metabolism. Bodor's work on these design concepts developed during the late 1970s and early 1980s, and came to prominence during the mid-1990s. Loteprednol etabonate, a soft corticosteroid designed and patented[35][36] by Bodor received final Food and Drug Administration (FDA) approval in 1998 as the active ingredient of two ophthalmic preparations (Lotemax and Alrex), currently the only corticosteroid approved by the FDA for use in all inflammatory and allergy-related ophthalmic disorders. Its safety for long-term use[37] further supports the soft drug concept, and in 2004, loteprednol etabonate[38][39][40] was also approved as part of a combination product (Zylet). A second generation of soft corticosteroids such as etiprednol dicloacetate[41] is in development for a full spectrum of other possible applications such as nasal spray for rhinitis or inhalation products for asthma.
The soft drug concept ignited research work in both academic (e.g., Aston University, Göteborg University, Okayama University, Uppsala University, University of Iceland, University of Florida, Université Louis Pasteur, Yale University) and industrial (e.g., AstraZeneca, DuPont, GlaxoSmithKline, IVAX, Janssen Pharmaceutica, Nippon Organon, Novartis, ONO Pharmaceutical, Schering AG) settings. Besides corticosteroids, various other therapeutic areas have been pursued such as soft beta-blockers, soft opioid analgetics, soft estrogens, soft beta-agonists, soft anticholinergics, soft antimicrobials, soft antiarrhythmic agents, soft angiotensin converting enzyme (ACE) inhibitors, soft dihydrofolate reductase (DHFR) inhibitors, soft cancineurin inhibitors (soft immunosuppressants), soft matrix metalloproteinase (MMP) inhibitors, soft cytokine inhibitors, soft cannabinoids, soft Ca2+ channel blockers (see[34] for a recent review).
Following the introduction of the CDS concepts, work along those lines started in numerous pharmaceutical centers around the world, and brain-targeting CDSs were explored for many therapeutic agents such as steroids (testosterone, progestins, estradiol, dexamethasone), anti-infective agents (penicillins, sulfonamides), antivirals (acyclovir, trifluorothymidine, ribavirin), antiretrovirals (AZT, ganciclovir), anticancer agents (Lomustine, chlorambucil), neurotransmitters (dopamine, GABA), nerve growth factor (NGF) inducers, anticonvulsants (Phenytoin, valproate, stiripentol), Ca2+ antagonists (felodipine), MAO inhibitors, NSAIDs and neuropeptides (tryptophan, Leu-enkephalin analogs, TRH analogs, kyotorphin analogs). A number of new chemical entities (NCE) were developed based on these principles, such as E2-CDS (Estredox[29] or betaxoxime[30] are in advanced clinical development phases.
A review of ongoing research using the general retrometabolic design approaches is conducted biennially at the Retrometabolism Based Drug Design and Targeting Conference, an international series of symposia developed and organized by Nicholas Bodor. Proceedings of each conference held have been published in the international pharmaceutical journal Pharmazie. Past conferences, and their published proceedings are:
- May 1997, Amelia Island, Florida; Pharmazie 52(7) S1, 1997
- May 1999, Amelia Island, Florida; Pharmazie 55(3), 2000
- May 2001, Amelia Island Florida; Pharmazie 57(2), 2002
- May 2003, Palm Coast, Florida; Pharmazie 59(5), 2004
- May 2005, Hakone, Japan; Pharmazie 61(2), 2006
- June 2007, Göd, Hungary; Pharmazie 63(3), 2008
- May 2009, Orlando, Florida; Pharmazie 65(6), 2010
- June 2011, Graz, Austria; Pharmazie 67(5), 2012
- May 2013, Orlando, Florida; Pharmazie 69(6), 2014
References
- ↑ Bodor N, Buchwald P (2012). Retrometabolic drug design and targeting. Hoboken, N.J.: John Wiley Sons. p. 418. ISBN 978-0-470-94945-0.
- ↑ Bodor, N.; Buchwald, P. (1999). "Recent advances in the brain targeting of neuropharmaceuticals by chemical delivery systems". Adv. Drug Deliv. Rev. 36: 229–254. doi:10.1016/s0169-409x(98)00090-8.
- ↑ Miller, G. (2002). "Breaking down barriers". Science. 297: 1116–1118. doi:10.1126/science.297.5584.1116.
- 1 2 3 Regier, D.A.; Boyd, J.H.; Burke, J.D., Jr.; Rae, D.S.; Myers, J.K.; Kramer, M.; Robins, L.N.; George, L.K.; Karno, M.; Locke, B.Z. (1988). "One-month prevalence of mental disorders in the United States". Arch. Gen. Psychiatry. 45: 977–986. doi:10.1001/archpsyc.1988.01800350011002. Cite uses deprecated parameter
|coauthors=(help) - ↑ Crone; Thompson, A.M. (1970). Crone, C.; Lassen, N.A., eds. Capillary Permeability: The Transfer of Molecules and Ions Between Capillary Blood and Tissue (C. ed.). Munksgaard: Copenhagen, Denmark. pp. 447–453.
- ↑ Oldendorf, W.H. (1974). "Lipid solubility and drug penetration of the blood-brain barrier". Soc. Exp. Biol. Med. 147: 813–816. doi:10.3181/00379727-147-38444.
- ↑ Rapoport, S.I. (1976). Blood-Brain Barrier in Psychology and Medicine. Raven Press, New York.
- ↑ Bradbury, M. (1979). The Concept of a Blood-Brain Barrier. Wiley, New York.
- ↑ Bodor, N.; Brewster, M.E. (1983). "Problems of delivery of drugs to the brain". Pharmacol. Ther. 19: 337–386. doi:10.1016/0163-7258(82)90073-0.
- ↑ Fenstermacher, J.D.; Rapoport, S.I. (1984). "The blood-brain barrier. In Microcirculation, Part 2". In Renkin, E.M.; Michel, C.C. Handbook of Physiology, Section 2: The Cardiovascular System, Vol. 4. American Physiological Society, Bethesda, MD. pp. 969–1000.
- ↑ Goldstein, G.W.; Betz, A.L. (1986). "The blood-brain barrier". Sci. Am. 255 (3): 74–83. doi:10.1038/scientificamerican0986-74.
- ↑ Bradbury, M.W.B., ed. (1992). Physiology and Pharmacology of the Blood-Brain Barrier, Handbook of Experimental Pharmacology, Vol. 103. Springer, Berlin.
- ↑ Begley, D.J. (1996). "The blood-brain barrier: principles for targeting peptides and drugs to the central nervous system". J. Pharm.Pharmacol. 48: 136–146. doi:10.1111/j.2042-7158.1996.tb07112.x.
- ↑ Schlossauer, B.; Steuer, H. (2002). "Comparative anatomy, physiology and in vitro models of the blood-brain and blood-retina barrier". Curr. Med. Chem. - Central Nervous System Agents. 2: 175–186.
- ↑ Betz, A.L.; Goldstein, G.W. "Brain capillaries: structure and function". In Lajtha, A. Structural Elements of the Nervous System; Handbook of Neurochemistry, Vol. 7. Plenum Press, New York. pp. 465–484.
- ↑ Pardridge, W.M. (1991). Peptide Drug Delivery to the Brain. Raven Press, New York.
- ↑ Abbott, N.J.; Bundgaard, M.; Cserr, H.F. (1986). "Comparative physiology of the blood-brain barrier". In Suckling, A.J.; Rumsby, M.G.; Bradbury, M.W.B. The Blood-Brain Barrier in Health and Disease. Ellis Horwood: Chichester, UK. pp. 52–72.
- ↑ Lo, E.H.; Singhal, A.B., Torchilin, V.P. and Abbott, N.J. (2001). "Drug delivery to damaged brain". Brain. Res. Rev. 38: 140–148. doi:10.1016/s0165-0173(01)00083-2. Cite uses deprecated parameter
|coauthors=(help) - ↑ Smith, Q.R. (1989). "Quantitation of blood-brain barrier permeability". In Neuwelt, E.A. Implications of the Blood-Brain Barrier and its Manipulation. Plenum Press, New York. pp. 85–113.
- ↑ Stewart, P.A.; Tuor, U.I. (1994). "Blood-eye barriers in the rat: Correlation of ultrastructure with function". J. Comp. Neurol. 340: 566–576. doi:10.1002/cne.903400409.
- ↑ Siegal, T.; Zylber-Katz, E. (2002). "Strategies for increasing drug delivery to the brain: focus on brain lymphoma.". Clin. Pharmacokinet. 41: 171–186. doi:10.2165/00003088-200241030-00002.
- ↑ Ehrlich, P. (1885). Das Sauerstoff Bedürfnis des Organismus: Eine farbenanalytische Studie (in German). Hirschwald, Berlin.
- ↑ Janzer, R.C.; Raff, M.C. (1987). "Astrocytes induce blood-brain barrier properties in endothelial-cells.". Nature. 325: 253–257. doi:10.1038/325253a0.
- 1 2 Bodor, N.; Buchwald, P. (2012). Retrometabolic Drug Design and Targeting. John Wiley & Sons, New York. ISBN 978-0-470-94945-0.
- ↑ Bodor, N.; Shek, E.; Higuchi, T. (1975). "Delivery of a quaternary pyridinium salt across the blood-brain barrier by its dihydropyridine derivative". Science. 190: 155–156. doi:10.1126/science.1166305.
- ↑ Bodor, N.; Farag, H.H.; Brewster, M.E. (1981). "Site-specific, sustained release of drugs to the brain". Science. 214: 1370–1372. doi:10.1126/science.7313698.
- ↑ Bodor, N.; Simpkins, J.W. (1983). "Redox delivery system for brain-specific, sustained release of dopamine". Science. 221: 65–67. doi:10.1126/science.6857264.
- ↑ Thomas, P. (1993). "Top ten advances of 1992". Harvard Health Letter. 18: 1–4.
- 1 2 Bodor, N.; Buchwald, P. (2006). "Brain-targeted delivery of estradiol: therapeutic potential and results obtained with a chemical delivery system approach". Am. J. Drug Deliv. 4: 161–175. doi:10.2165/00137696-200604030-00004.
- 1 2 Bodor, N.; Buchwald, P. (2005). "Ophthalmic drug design based on the metabolic activity of the eye: soft drugs and chemical delivery systems". AAPS J. 7: article 79 (E820–E833).
- ↑ Bodor, N. (1977). "Novel approaches for the design of membrane transport properties of drugs". In Roche, E.B. Design of Biopharmaceutical Properties through Prodrugs and Analogs. Academy of Pharmaceutical Sciences; Washington, D.C. pp. 98–135.
- ↑ Bodor, N. (1982). "Designing safer drugs based on the soft drug approach". Trends Pharmacol. Sci. 3: 53–56. doi:10.1016/0165-6147(82)91008-2.
- ↑ Bodor, N. (1984). "Novel approaches to the design of safer drugs: Soft drugs and site-specific chemical delivery systems". Adv. Drug Res. 13: 255–331.
- 1 2 Retrometabolism-based drug design and targeting (2010). "3". In Abraham, D. Burger's Medicinal Chemistry, Drug Discovery and Development; Vol. 2: Discovering Lead Molecules. Wiley & Sons; New York.
- ↑ Bodor, N. (1981). "Stéroïds doux exerçant une activité anti-inflammatoire. (Steroids having anti-inflammatory activity)". Belgian Patent BE889,563 (in French). Intl. Classif. C07J/A61K.
- ↑ Bodor, N. (1991). "Soft Steroids having anti-inflammatory activity". U.S. Patent 4,996,335.
- ↑ Ilyas, H.; Slonim, C.B.; Braswell, G.R.; Favetta, J.R.; Schulman, M. (2004). "Long-term safety of loteprednol etabonate 0.2% in the treatment of seasonal and perennial allergic conjunctivitis". Eye Contact Lens. 30: 10–13. doi:10.1097/01.icl.0000092071.82938.46.
- ↑ Bodor, N.; Buchwald, P. (2002). "Design and development of a soft corticosteroid, loteprednol etabonate". In Schleimer, R.P.; O'Byrne, P.M.; Szefler, S.J.; Brattsand, R. Inhaled Steroids in Asthma. Optimizing Effects in the Airways. Lung Biology in Health and Disease, Vol. 163. Marcel Dekker, New York. pp. 541–564.
- ↑ Hermann, R.; Locher, M.; Siebert-Weigel, M.; LaVallee, N.; Derendorf, H. and Hochhaus, G. (2004). "Intranasal loteprednol etabonate in healthy male subjects: pharmacokinetics and effects on endogenous cortisol". J. Clin. Pharmacol. 44: 510–519. doi:10.1177/0091270004264163.
- ↑ Bodor, N.; Buchwald, P. (2006). "Corticosteroid design for the treatment of asthma: structural insights and the therapeutic potential of soft corticosteroids". Curr. Pharm. Des. 12: 3241–3260. doi:10.2174/138161206778194132.
- ↑ Bodor, N. (1999). "Androstene Derivatives". U.S. Patent 5,981,517.