Resolution by Proxy
| Content | |
|---|---|
| Description | Calculates structure resolution using coordinate data only |
| Contact | |
| Research center | University of Alberta |
| Laboratory | Dr. David Wishart |
| Primary citation | [1] |
| Access | |
| Data format | Data Input: X-ray or NMR coordinates (PDB format); Data Output: Estimated resolution in Angstroms |
| Website | http://www.resprox.ca; http://www.resprox.ca/download.html |
| Miscellaneous | |
| Data release frequency | Every 1-2 years with periodic corrections and updates |
| Curation policy | Manually curated |
Resolution by Proxy (ResProx) is a method for assessing the equivalent X-ray resolution of NMR-derived protein structures. ResProx calculates resolution from coordinate data rather than from electron density or other experimental inputs. This makes it possible to calculate the resolution of a structure regardless of how it was solved (X-ray, NMR, EM, modeling, ab initio prediction). ResProx was originally designed to serve as a simple, single-number evaluation that allows straightforward comparison between the quality/resolution of X-ray structures and the quality of a given NMR structure. However, it can also be used to assess the reliability of an experimentally reported X-ray structure resolution, to evaluate protein structures solved by unconventional or hybrid means and to identify fraudulent structures deposited in the PDB.[1] ResProx incorporates more than 25 different structural features to determine a single resolution-like value. ResProx values are reported in Angstroms. Tests on thousands of X-ray structures show that ResProx values match very closely to resolution values reported by X-ray crystallographers.[1] Resolution-by-proxy values can be calculated for newly determined protein structures using a freely accessible ResProx web server.[1] This server accepts protein coordinate data (in PDB format) and generates a resolution estimate (in Angstroms) for that input structure.
Background and Rationale
In X-ray crystallography, resolution is a measure of the resolvability or precision in the electron density map of a molecule. Resolution is usually reported in Angstroms (10–10 meters) for X-ray crystal structures. The smaller the number, the better the degree of atomic resolution. In protein X-ray crystallography the best resolution typically attainable is about 1 Angstrom. This level of resolution allows individual hydrogen atoms to be visualized and heavy atoms (C, O, N) to be very accurately mapped. Most protein structures solved today have a resolution of 1.5 to 2.5 Angstroms, which means the hydrogen atoms are not visible and there is some uncertainty in the precise location of the heavy atoms. Protein structures with a resolution of >2.5 Angstroms generally have a number of coordinate inaccuracies as well as other structural problems. When the resolution is greater than 3.5 Angstroms, there is often considerable uncertainty in both the atom locations and even the identity of individual amino residues. In other words, resolution is inversely correlated with structure quality (i.e. higher numbers mean poorer structures). This trend in protein structure quality for X-ray resolution matches very closely to the trend seen the quality of NMR-determined protein structures. Some NMR structures have large numbers of constraints (NOEs, H-bonds, J-couplings, dipolar couplings), excellent geometry, high structure quality and very tight ensembles with excellent atomic precision (RMSDs < 1 Angstrom). Other NMR structures have very few constraints, poor geometry or poor structure quality and very loose ensembles (RMSDs > 3 Angstroms). However, there is no simple mapping between NMR RMSD values and X-ray resolution values. That is, an NMR ensemble with 1 Angstrom RMSD does not correspond in quality or precision to an X-ray structure with 1 Angstrom resolution. This is because the RMSD measure is both a function of the number of structures used in the ensemble and the selection bias of the spectroscopist who desposits the structural ensemble. Likewise, in NMR it is possible to generate high quality, precisely determined protein structures using relatively few, well-chosen constraints. It is also possible to generate very low quality NMR structures from large numbers of carelessly assessed, mistaken or mis-assigned constraints.
Over the past 20 years several methods have been proposed to calculate “equivalent resolution” using only X-ray coordinate data (rather than X-ray diffraction data). Some were designed specifically for evaluating NMR structures such as Procheck-NMR [2] while others were designed more for structure quality evaluation and validation of X-ray structures such as MolProbity,[3] and RosettaHoles2.[4] However, these methods rely on a relatively small number of protein structure quality measures to predict resolution (4, 3, and 1 measures, respectively) and consequently the correlation between observed (X-ray) resolution and the predicted resolution is not particularly good. By expanding the number of structure features to include the distribution of torsion angles, the presence of atom clashes, the normality of hydrogen bonding, the numbers of violations of bond lengths and bond angles, the presence of cavities, residue-specific packing volumes, packing efficiency and threading energies it is possible to improve this correlation quite substantially.
The ResProx Algorithm
ResProx uses a collection of 25 different protein structure features (such as torsion angle distributions, hydrogen bonding, packing volume, cavities, Molprobity measures) that were used in a Support Vector Regression method to maximize the correlation between the predicted resolution and the observed X-ray resolution on a set of 2400 protein structures with known X-ray resolution. The exact details of the algorithm are provided in a paper published by Dr. Wishart and colleagues.[1] After training and appropriate validation on independent tests sets, this SVR model is able to estimate the resolution of solved X-ray structures with a correlation coefficient of 0.92, mean absolute error of 0.28 Angstroms. This is about 15-30% better than existing methods. This is shown in Figure 1. Because the performance of the ResProx method is so high and because it only needs coordinate data to generate an estimate of the equivalent X-ray resolution, it is ideally suited to be applied to NMR structures. Interestingly when NMR structures are analyzed by ResProx, the average NMR structure has an equivalent X-ray resolution of 2.8 Angstroms—which is relatively poor (Fig. 2). This is in agreement with qualitative observations regarding the overall quality and precision of NMR structures. As seen in Figure 2, a very small number NMR structures exhibit a resolution equivalent to < 1.0 Angstroms—but these are rare.
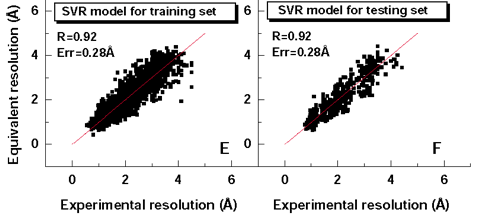
Figure 1. Performance of ResProx against training and testing data.

Figure 2. Histogram of ResProx equivalent resolution for NMR models and experimental resolution for X-ray structures. 500 NMR ensembles and 500 X-ray structures were randomly selected from the PDB. Proteins were grouped in 0.25Å resolution bins. Resolution values on the X-axis indicate the upper limit of each resolution bin. Values for NMR structures and X-ray structures represent the number of structures in each resolution bin.
The ResProx Server
The ResProx web server a freely accessible server that accepts NMR protein coordinate data (in PDB format) and generates a resolution estimate (in Angstroms) for that NMR structure. A downloadable version of ResProx is also available. ResProx also provides a list of List of 50834 protein structures with PDB identifiers along with their observed resolution and corresponding ResProx values.
References
- 1 2 3 4 5 Berjanskii, M; Zhou J; Liang Y; Li G; Wishart DS (July 2012). "Resolution-by-proxy: a simple measure for assessing and comparing the overall quality of NMR protein structures.". J Biomol NMR. 53 (3): 167–80. doi:10.1007/s10858-012-9637-2. PMID 22678091.
- ↑ Laskowski, RA; Rullmannn JA; MacArthur MW; Kaptein R; Thornton JM (1996). "AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR.". J Biomol NMR. 8 (4): 477–486. doi:10.1007/bf00228148. PMID 9008363.
- ↑ Chen, VB; Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010). "MolProbity: all-atom structure validation for macromolecular crystallography.". Acta Crystallogr D. 66 (Pt 1): 12–21. doi:10.1107/S0907444909042073. PMC 2803126
 . PMID 20057044. Cite uses deprecated parameter
. PMID 20057044. Cite uses deprecated parameter |coauthors=(help) - ↑ Sheffler, W; Baker D (2010). "RosettaHoles2: a volumetric packing measure for protein structure refinement and validation.". Protein Sci. 19 (10): 1991–1995. doi:10.1002/pro.458. PMC 2998733. PMID 20665689.