Randle cycle
The Randle cycle, also known as the glucose fatty-acid cycle, is a metabolic process involving the competition of glucose and fatty acids for substrates.[1] It is theorized to play a role in explaining type 2 diabetes and insulin resistance.[2][3]
It was named for Philip Randle, who described it in 1963.[4]
Cycle
The Randle cycle is a biochemical mechanism involving the competition between glucose and fatty acids for their oxidation and uptake in muscle and adipose tissue. The cycle controls fuel selection and adapts the substrate supply and demand in normal tissues. This cycle adds a nutrient-mediated fine tuning on top of the more coarse hormonal control on fuel metabolism. This adaptation to nutrient availability applies to the interaction between adipose tissue and muscle. Hormones that control adipose tissue lipolysis affect circulating concentrations of fatty acids, these in turn control the fuel selection in muscle. Mechanisms involved in the Randle Cycle include allosteric control, reversible phosphorylation and the expression of key enzymes.[5] The energy balance from meals composed of differing macronutrient composition is identical, but the glucose and fat balances that contribute to the overall energy balance change reciprocally with meal composition.[6]
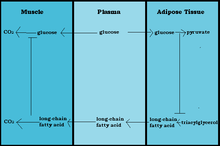
Glucose is Spared and Rerouted
Fasted State
When fasting, the activation of lipolysis provides fatty acids as the preferred fuel source for respiration. In the liver β-oxidation of fatty acids fulfills the local energy needs and may lead to ketogenesis (creating ketone bodies out of fatty acids.) The ketone bodies are then used to meet the demands of tissues other than the liver. The inhibition of glucose oxidation causes fatty acids and ketone bodies to contribute to a glucose-sparing effect, which is an essential survival mechanism for the brain during times of starvation. This inhibition of glucose oxidation at the level of pyruvate dehydrogenase preserves pyruvate and lactate, both of which are gluconeogenic precursors.[5]
Fed State
The glucose fatty acid cycle is also observed in the fed state after a high-fat meal or during exercise. This is when plasma concentrations of fatty acids or ketone bodies are increased. The glucose that is not oxidized is then rerouted to glycogen. This rerouting to glycogen explains the rapid resynthesis of muscle glycogen after exercise as well as the increased glycogen content in muscles found in starvation or diabetes. This mechanism replenishes the intermediates of the citric acid cycle.[5]
Inhibition of Glycolytic Pathway
The impairment of glucose metabolism by fatty acid oxidation is mediated by the short-term inhibition of several glycolytic processes. The extent of inhibition increases along the glycolytic pathway, being most severe at the level of pyruvate dehydrogenase and less severe at the level of glucose uptake and 6-phosphofructo-1-kinase (PFK-1).[5] This sequence occurs because of the initial event, triggered by fatty acid oxidation, is an increase in the mitochondrial ratios of [acetyl-CoA]/[CoA] and [NADH]/[NAD+]. These both serve to inhibit pyruvate dehydrogenase activity.[7] It has been proposed that these changes lead to an accumulation of cytosolic citrate, which in turn inhibits PFK-1, followed by an increase in glucose 6-phosphate, which eventually inhibits hexokinase.[5]

Hemodynamic Stress
Hemodynamic stress overrides fatty acid inhibition of glucose metabolism. During this time there is a decrease in substrate supply and an increase in the substrate demand. This leads to an activation of AMP-activated protein kinase (AMPK) as the AMP concentration rises in intracellular fluids and the ATP concentration decreases. The stress-induced activation of AMPK provides an immediate metabolic adaption and protects the heart from ischemic stress.[5][8][9]
Fatty Acid Oxidation Inhibition by Malonyl-CoA
Malonyl-CoA signals glucose utilization and it controls the entry and oxidation of long-chain fatty acids (LCFA) in the mitochondria. Circulating glucose in the liver stimulates its uptake. Glucose oxidation produces citrate which can be converted to malonyl-CoA by acetyl-CoA carboxylase. Malonyl-CoA inhibits the carnitine palmitoyltransferase (CPT) that controls the entry and oxidation of LCFA. The glucose-derived malonyl-CoA prevents the oxidation of fatty acids and favors fatty acid esterification.[4][5]

Cytosolic events controlling fatty acid oxidation
Malonyl-CoA concentration
The concentration of malonyl-CoA depends on the balance between acetyl-CoA carboxylas (ACC) and malonyl-CoA decarboxylase (MCD). AMP-activated protein kinase (AMPK) is reported to phosphorylate and inactivate liver ACC. This in turn decreases malonyl-CoA concentrations which stimulates fatty acid oxidation and ketogenesis by glucagon in the liver. AMPK phosphorylates and inactivates ACC in the liver and other tissues.[4][5]
Integration of AMPK and ACC in the glucose-fatty acid cycle
Inhibition of fatty acid oxidation requires that ACC is active. Both AMPK and MCD are inactive and glucose uptake is stimulated. The LCFAs are then rerouted to esterification.[10] These conditions exist in tissues rich in oxygen, in which AMPK is inactive and glucose inactivates the AMPK (researched in skeletal muscle).[11]
The inhibition of MCD suppresses the oxidation of fatty acids and stimulates glucose oxidation. In a study on MCD deficient mice there was no difference in the oxidation of fatty acids and glucose in the heart under aerobic conditions. It is theorized that the overexpression of fatty acids being used makes up for the lack of MCD.[12]
Fatty Acid Uptake
Long chain fatty acid uptake is mediated by several transporters, including FAT (fatty acid translocase)/CD36. CD36 deletion rescues lipotoxic cardiomyopathy. FAT/CD36 may be controlled by insulin and AMPK. Increased transport coupled to the formation of the CoA derivatives and the resulting AMPK activation should ensure efficient fatty acid uptake and metabolism.[5]
Mitochondrial Events Controlling Fuel Selection
Fatty acids are preferentially oxidized because of the inactivation of PDH by fatty acid oxidation inhibiting glucose oxidation. This suggests that mitochondrial metabolism may control fuel selection. Cellular respiration is stimulated by fatty acids and this relates to an increase in the mitochondrial NADH to NAD+ ratio, suggesting that energy provision overtakes energy consumption. Switching from glucose to fatty acid oxidation leads to a bigger proportion of electrons being transported to complex 2 rather than complex 1 of the respiratory chain. This difference leads to a less efficient oxidative phosphorylation. By oxidizing fatty acids, mitochondria increase their respiration while increasing the production of ROS.[5]
Fatty Acids and Insulin
Fatty acids may act directly upon the pancreatic β-cell to regulate glucose-stimulated insulin secretion. This effect is biphasic. Initially fatty acids potentiate the effects of glucose. After prolonged exposure to high fatty acid concentrations this changes to an inhibition.[13] Randle suggested that the term fatty acid syndrome would be appropriate to apply to the biochemical syndrome resulting from the high concentration of fatty acids and the relationship to abnormalities of carbohydrate metabolism, including starvation, diabetes and Cushing’s syndrome.[4]
References
- ↑ Bevilacqua S, Buzzigoli G, Bonadonna R, et al. (1990). "Operation of Randle's cycle in patients with NIDDM". Diabetes. 39 (3): 383–9. doi:10.2337/diabetes.39.3.383. PMID 2307295.
- ↑ Shuldiner AR, McLenithan JC (2004). "Genes and pathophysiology of type 2 diabetes: more than just the Randle cycle all over again". J. Clin. Invest. 114 (10): 1414–7. doi:10.1172/JCI23586. PMC 525752
 . PMID 15545992.
. PMID 15545992. - ↑ Delarue J, Magnan C (2007). "Free fatty acids and insulin resistance". Current opinion in clinical nutrition and metabolic care. 10 (2): 142–8. doi:10.1097/MCO.0b013e328042ba90. PMID 17285001.
- 1 2 3 4 Randle PJ, Garland PB, Hales CN, Newsholme EA (1963). "The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus". Lancet. 1 (7285): 785–9. doi:10.1016/S0140-6736(63)91500-9. PMID 13990765.
- 1 2 3 4 5 6 7 8 9 10 Hue L, Taegtmeyer H (2009). "The Randle cycle revisited: a new head for an old hat". American Journal of Physiology. Endocrinology and Metabolism. 297: E578–E591. doi:10.1152/ajpendo.00093.2009.
- ↑ Frayn K.N. (2003). "The glucose-fatty acid cycle: a physiological perspective". Biochem Soc Trans. 31 (Pt 6): 1115–9. doi:10.1042/bst0311115. PMID 14641007.
- ↑ Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998). "Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex". Biochem J. 329: 191–6. PMC 1219031. PMID 9405293.
- ↑ Kudo N, Gillespie JG, Kung L, Witters LA, Schulz R, Clanachan AS, Lopaschuk GD (1996). "Characterization of 5'AMP-activated protein kinase activity in the heart and its role in inhibiting acetyl-CoA carboxylase during reperfusion following ischemia". Biochim Biophys Acta. 1301 (1-2): 67–75. doi:10.1016/0005-2760(96)00013-6. PMID 8652652.
- ↑ Goodwin GW, Taegtmeyer H (2000). "Improved energy homeostasis of the heart in the metabolic state of exercise". American Journal of Physiology. Heart and Circulatory Physiology. 279: H1490–H1501.
- ↑ Clark H, Carling D, Saggerson D (2004). "Covalent activation of heart AMP-activated protein kinase in response to physiological concentrations of long-chain fatty acids". Eur J Biochem. 271 (11): 2215–24. doi:10.1111/j.1432-1033.2004.04151.x. PMID 15153111.
- ↑ Itani SI; Saha AK; Kurowski TG; Coffin HR; Tornheim K; Ruderman NB (2003). "Glucose Autoregulates Its Uptake in Skeletal Muscle-Involvement of AMP-Activated Protein Kinase". Diabetes. 52: 1635–1640. doi:10.2337/diabetes.52.7.1635.
- ↑ Dyck JRB, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M, Kawase Y, JishageK, Lopaschuk GD (2006). "Absence of Malonyl Coenzyme A Decarboxylase in Mice Increases Cardiac Glucose Oxidation and Protects the Heart From Ischemic Injury". Journal of the American Heart Association. 114: 1721–1728. doi:10.1161/CIRCULATIONAHA.106.642009.
- ↑ Grill V, Qvigstad E (2000). "Fatty acids and insulin secretion". British Journal of Nutrition. 83: S79–S84. doi:10.1017/S0007114500000994.