Illumina Methylation Assay
The Illumina Methylation Assay using the Infinium I platform uses 'BeadChip' technology to generate a comprehensive genome wide profiling of human DNA methylation. Similar to bisulfite sequencing and pyrosequencing, this method quantifies methylation levels at specific loci within the genome. This assay is used for methylation probes on the Illumina 27k methylation array. Probes on the 27k array target regions of the human genome to measure methylation levels at 27,578 CpG dinucleotides in 14,495 genes.[1]
Background
DNA methylation plays a significant role in the epigenetic regulation of chromatin structure, which in the last decade has been recognized to be important in the regulation of gene expression, development and genetic imprinting in vertebrates.[1] Changes in the methylation pattern and level have been shown to contribute to cancer and various developmental diseases.[2] For example, hypermethylation at the promoter CpG islands of a tumour suppressor gene, which in turn leads to its silencing, is frequently associated with tumourgenesis.[2] A large scale measurement of DNA methylation patterns from a wide selection of genes may enable us to understand better the relationships between epigenetic changes and the genesis of different diseases and a better understanding of the role that epigenetics plays in tissue specific differentiation.
Material
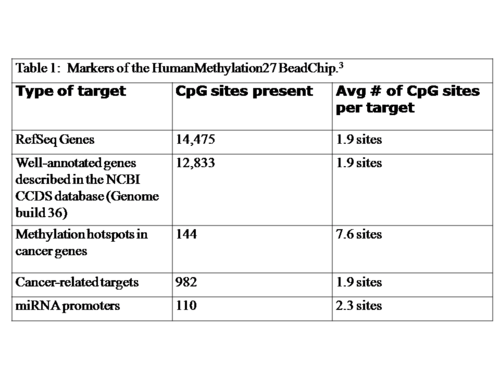
The chip contains 27,578 individual CpG sites, spread across 14,495 genes.[1] These genes include RefSeq genes from the NCBI CCDS Database, cancer genes that show differential methylation patterns during their course of progression and microRNA promoters.[3] The markers included in the chip are summarized in Table 1.[3]
Method
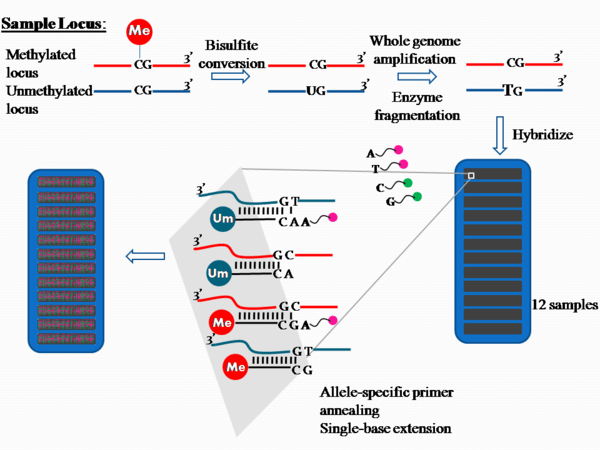
The process is outlined in Figure 1.
Bisulfite treatment
Approximately 1 ug of genomic DNA is used in bisulfite conversion to convert the unmethylated cytosine into uracil. The product contains unconverted cytosine where they were previously methylated, but cytosine converted to uracil if they were previously unmethylated.
Whole genomic DNA amplification
The bisulfite treated DNA is subjected to whole genome amplification (WGA) via random hexamer priming and Phi29 DNA polymerase, which has a proofreading activity resulting in error rates 100 times lower than the Taq polymerase. The products are then enzymatically fragmented,[1] purified from dNTPs, primers and enzymes, and applied to the chip.[4]
Hybridization and Single-base extension
On the chip, there are two bead types for each CpG site per locus. Each locus tested is differentiated by different bead types, there are over 200,000 bead types available.[1] Each of the bead types are attached to a single stranded 50-mer DNA oligonucleotides that differ in sequence only at the free end; this type of probe is known as an Allele specific oligonucleotide. One of the bead types will correspond to the methylated cytosine locus and the other will correspond to the unmethylated cytosine locus, which has been converted into uracil during bisulfite treatment and later amplified as thymine during whole genome amplification.
The bisulfite converted amplified DNA products are denatured into single strands and hybridized to the chip via allele specific annealing to either the methylation specific probe or the non-methylation probe. Hybridization is followed by single-base extension with hapten labelled dideoxynucleotides. The ddCTP is labelled with biotin while ddATP, ddUTP and ddGTP are labelled with 2,4-dinitrophenol (DNP) [5]
Fluorescence staining and scanning of chip
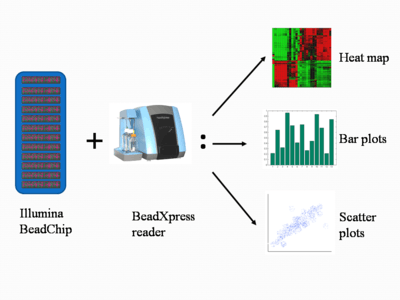
After incorporation of these hapten labelled ddNTPs, multi-layered immunohistochemical assays are performed by repeated rounds of staining with a combination of antibodies to differentiate the two types.[5] After staining, the chip is scanned to show the intensities of the unmethylated and methylated bead types. (Figure 2).[1] The raw data are analyzed by the proprietary software, and the fluorescence intensity ratios between the two bead types are calculated. A ratio value of 0 equals to non-methylation of the locus; a ratio of 1 equals to total methylation; a value of 0.5 means that one copy is methylated and the other is not, in the diploid human genome.[1][3]
Analysis of methylation data
The scanned microarray images of methylation data are further analyzed by the system, which normalizes the raw data to reduce the effects of experimental variation, background and average normalization, and performs standard statistical tests on the results.[6] The data can then be compiled into several types of figures for visualization and analysis. Scatter plots are used to correlate the methylation data; bar plots to visualize relative levels of methylation at each site tested; heat maps to cluster the data to compare the methylation profile at the sites tested. Figure 2 shows the different types of results generated.
Advantages and disadvantages
Note: this Advantages-Disadvantages section is appropriate as it describes the relative technical merits of a process and should not be contorted into a seemingly neutral equivalent. This "Pro-and-Con" is not a value judgment. It is a well-established way of elucidating technical applicability in science and engineering. - op ed.
Advantages
- No PCR is required, which means that there will be no selective bias towards shorter fragments.[3]
- Ability to survey up to 12 samples per chip allows for high throughput processing.[3]
- Allows integration of data between other platforms such as gene expression and microRNA profiling.[3]
- The method looks at ~2 CpG sites per CpG island, providing genome-wide coverage of methylation patterns
Disadvantages
- Not every gene annotated in the NCBI database was included in the design of this assay, which covers 14,495 genes out of the 17,052 GeneIDs present to date (Human build 36.3).[7]
- According to Staaf et al. (2008),[8] “the Infinium II assay seemed to have dye intensity biases between the two channels used in fluorescence detection. Furthermore, this bias was not eliminated even after the data had gone through normalization algorithms used in the BeadStudio software.” This concern, while valid for the GoldenGate methylation assay, is not relevant to the HumanMethylation27k chips, where both probes in a pair extend and fluoresce within the same channel.[8]
See also
- MethDB DNA Methylation Database
External links
- Epigenetics Methylation Station
- Nature Reviews:DNA methylation Collection in Nature Reviews
- DNA/Methylation Analysis Protocols
- Illumina, Infinium Methylation
- OMICtools: an educational directory for DNA methylation array data analysis.
References
- 1 2 3 4 5 6 7 Weisenberger, DJ. et al. (2008) Comprehensive DNA Methylation Analysis on the Illumina Infinium Assay Platform.
- 1 2 DNA methylation in development and human disease. Gopalakrishnan S, Van Emburgh BO, Robertson KD. Mutation Research. 2008 Dec 1;647(1-2):30-8. Epub 2008 Aug 20. Review.
- 1 2 3 4 5 6 Illumina: DNA Methylation Analysis: http://www.illumina.com/downloads/DNAMethylationAnalysis_DataSheet.pdf
- ↑ Gunderson, KL. et al. A genome-wide scalable SNP genotyping assay using microarray technology. Nature Genetics 37, 549 - 554 (2005).
- 1 2 Steemers, FJ. et al. Whole-genome genotyping with the single-base extension assay. Nature methods, Vol. 3, No. 1, Jan, 31 – 33 (2006).
- ↑ BeadStudio Methylation Module User Guide. Version 3. Illumina Systems & Software. 2006
- ↑ NCBI, Consensus CDS (CCDS) project
- 1 2 Staaf, J. et al. Normalization of Illumina Infinium whole-genome SNP data improves copy number estimates and allelic intensity ratios. BMC Bioinformatics 2008, 9:409.