History and naming of human leukocyte antigens
Human leukocyte antigens (HLA) began as a list of antigens identified as a result of transplant rejection. The antigens were initially identified by categorizing and performing massive statistical analyses on interactions between blood types.[1] This process is based upon the principle of serotypes. HLA are not typical antigens, like those found on surface of infectious agents. HLAs are alloantigens, they vary from individual to individual as a result of genetic differences. An organ called the thymus is responsible for ensuring that any t-cells that attack self proteins are not allowed to live. In essence, every individual's immune system is tuned to the specific set of HLA and self proteins produced by that individual. (For more information please see immune tolerance). Where this goes wrong is when tissues are transferred to another person. Since individuals almost always have different "banks" of HLAs, the immune system of the recipient recognizes the transplanted tissue as non-self and destroys the offending tissue. It was through the realization of this that HLAs were discovered.
Discovery
The thought that the mammalian body must have some way of identifying introduced foreign particles first arose during World War II. Numerous theories were proposed and it wasn't until 1958 that the first of these identifying proteins was found.[2] The first standardized naming system was established in 1968 by the WHO Nomenclature Committee for Factors of the HLA System.[3] HLA research didn't heat up until the 1980s when a group of researchers finally elucidated the shape of the HLA-A*02 protein (just one of many specific HLA proteins).[1] Even more recently, in 2010, the WHO committee responsible for naming all HLA proteins revised their standards for naming to introduce more clarity and specificity in the naming system.[3] It started with a plane crash in the height of the London Blitz. The pilot suffered severe burns requiring skin grafts. However, at the time skin grafts were a risky business, often being rejected for unknown reasons.[1] The search for a reason spanned the next four decades and in some areas is still continuing today.

Identification of Non-Self
Peter Medawar was a zoologist turned clinician, who specialized in burn trauma. A plane crash near his home changed the path of his career, turning his work with burns from mere academia to a full on quest to save lives. Medawar and a Scottish surgeon, Tom Gibson, were tasked with working the Burns Unit of the Glasgow Royal Infirmary. The first insight came when the pair decided to experiment, and grafted part of a wound with the patient's skin, and another part with skin from the patient's brother. Within days the skin grafts from the brother were completely destroyed. Successive skin grafts from the brother were destroyed even faster, a fact that gave them the evidence they needed to implicate the immune system. Medawar later repeated this experiment on rabbits and 625 surgeries later validated their initial conclusions.[4] Medawar then set out in search of the reason why rabbits rejected non-self grafts.[1]
Medawar continued his work, this time with a team of three at the University College London during the 1950s. Medawar's coworkers were Leslie Brent, a PhD student, and Rupert Billingham, Medawar's first graduate student at Oxford several years prior. Through carefully planned experimentation, the trio showed that mice exposed to cells of unrelated mice as fetuses did not reject skin grafts from those same mice.[5] For this discovery, Medawar and Australian scientist Macfarlane Burnet earned the 1960 Nobel Prize.[1]

Learned Self Tolerance
Burnet, independently of Medawar, came to the conclusion that the immune system must learn to tolerate any self cells, and hypothesized that this must occur during fetal development. For this, he jointly was awarded the Nobel Prize in 1960. Burnet's work continued and in 1957 along with Niels Jerne published a paper that modified and revolutionized antibody theory. "Burnet speculated that one cell makes one particular shape of antibody and that all our antibody-making immune cells together make an unimaginably vast repertoire of 10 billion antibodies, each having a slightly different shape".[6] Thus, whenever a non-self molecule appears in the human body, one of these antibodies will have an accurate enough shape to bind to that molecule. This idea is known as Clonal Selection Theory. At the time, many leading scientists including Linus Pauling and James Watson completely rejected the idea, but repeated experimentation intended to disprove the theory actually served to build up a large body of evidence supporting Burnet and Jerne's theory.[1]
The biggest weakness in Burnet's theory was that he had no explanation for how the body selected for immune cells that only identified non-self. In 1961, Jacques Miller published a paper offering an explanation. Miller was a PhD student at the Chester Beatty Research Institute in London. His discovery centered on the thymus. The thymus had long been regarded as nothing more than a repository for dead cells. Miller didn't buy this hypothesis. By removing the thymus of leukemic mice early in life, he found that the mice had a drastically weakened immune system. Taking inspiration from Medawar's skin transplant work, he performed a series of skin-graft experiments that showed that these immunocompromised mice didn't reject skin grafts from non-genetically identical mice. Miller then hypothesized that the thymus was essential in the construction and maintenance of the immune system. At this point Burnet came back into the picture, extending the hypothesis to specify that the dead cells found in the thymus are not any old immune cells, but instead the cells that are activated by self molecules. In other words, any molecule that binds to and hence "recognizes" a self molecule is killed before exiting the thymus. These cells were later found to be one of two types of immune cells, the T-cells (named for their origin, the thymus).[1]
Identification of the first HLAs
The discovery of the first HLA was very much a mystery. In 1958 Jean Dausset noticed that blood serum from one person could react with the white blood cells of another. He had no idea why, but he named the causative agent MAC. Around the same time other researchers were making similar discoveries. Rose Payne and Jon van Rood made an identical conclusion from observations of interactions between the blood of women who had been pregnant multiple times and the white blood cells of others. They hypothesized that this was because they had been "sensitized" (an immunological term meaning previously exposed to and thus more reactive towards) to the non-self proteins of the father through tissue damage during birth. At this point the researchers all realized that the sheer quantity of data they were capable of obtaining was vastly greater than that of any previous study and so collaboration would be essential. The first international meeting, in 1964, highlighted the difficulties of such massive collaborative work. Different experimental methods and inconsistency in the execution of the same tests and a non-homogeneity of naming systems added together to make collaboration incredibly difficult.
World Health Organization steps in
In 1967 the World Health Organization (WHO) decided that the HLA research needed an official naming system. This in turn would aid in organization and would more easily facilitate the unification of data being collected at numerous laboratories across the world. This committee is still in existence today and vastly accelerated the rate of HLA research. The first meeting of this committee in 1968 set forth guidelines and rules that govern HLAs. First, compatibility genes were divided into two types, class I and class II. Class I molecules were identified via reactions between blood serum and cells. Class II molecules were identified by mixtures of white blood cells. Second, the compatibility genes were renamed Human Leukocyte Antigens (HLA).[1] Despite this clarification and the ever increasing number of identified HLAs, nobody knew how they worked.
MHC Restriction

Late in 1973 a pair of researchers in Australia, Rolf Zinkernagel and Peter Doherty made a revelatory discovery that altered the thinking of immunologists forever. The pair was doing research on viral infections in mice and noticed that T-cells that prevented viral infections in some mice wouldn't always prevent the same infection in other mice. After looking at the MHCs present in the mice, they realized that cytotoxic T-cells could only identify virus infections in cells with the right Class I compatibility gene. Traditional thinking was that the immune system identified infections directly but this discovery turned that theory on its head. Compatibility genes were essential in immune system mediated viral clearing. The pair coined the term "MHC Restriction" to describe this relationship between T-cells, specific MHC proteins, and viral detection.[1] In 1975, in an article in the journal Lancet, they introduced the idea of "altered self", meaning that viruses alter the MHC proteins and this alteration is detected by T-cells.[8] For their work they won the 1996 Nobel Prize.[1] It took the work of many others to determine how T-cells made this identification.
Discovering the protein shape
Nearly all important molecules in the body are proteins. Proteins work by each having a specific sequence of amino acids and a specific shape. Determining the order of amino acids is relatively simple. Finding the shape requires the use of x-ray crystallography and is anything but easy.[9] It took a team of three researchers at Harvard, Don Wiley, Jack Strominger, and Pamela Bjorkman, eight years to ferret out the structure of the HLA protein. They worked specifically with HLA-A*02. Bjorkman did the majority of the leg work and in the seven years managed to piece together the structure of 90% of the protein. That last 10% was elusive though. It took another year of work to finally unveil the complete structure of HLA-A*02. They completed their work in the spring of 1987, discovering that the final 10% made a "cup" (of sorts) located on top of the molecule. It was the perfect size to hold peptides. Other researchers had previously determined that T-Cells can recognize cells infected with a virus, cells injected with a single protein from a virus, and even cells injected with pieces of protein from a virus. The discovery of the HLA protein structure made it starkly clear that the HLA proteins hold viral peptides in their binding groove. But the research team from Harvard wasn't done. They also observed that there was clearly a peptide in the binding groove of the HLA molecules they used to determine the shape. However, the cells they had extracted the protein from were definitely not infected by any disease causing viruses.[1] The conclusion they made and the conclusion that has stuck to this day, is that HLA molecules can bind both self, and non-self peptides.
Nomenclature
Current HLA naming system
The most recent HLA naming system was developed in 2010 by the WHO Committee for Factors of the HLA System. There are two types of MHCs, Class I and Class II. Both are named using the same system. Currently there are 7,678 Class I alleles and 2,268 Class II alleles.
| HLA class I allele and protein quantities[10] | ||||||
|---|---|---|---|---|---|---|
| Gene | A | B | C | E | F | G |
| Alleles | 2432 | 3086 | 2035 | 13 | 22 | 50 |
| Proteins | 1740 | 2329 | 1445 | 5 | 4 | 16 |
| HLA Class I pseudogene alleles[10] | ||||||
|---|---|---|---|---|---|---|
| Gene | H | J | K | L | P | V |
| Alleles | 12 | 9 | 6 | 5 | 5 | 3 |
| HLA class II alleles and proteins[10] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | DRA | DRB | DQA1 | DQB1 | DPA1 | DPB1 | DMA | DMB | DOA | DOB |
| Alleles | 7 | 1476 | 51 | 459 | 37 | 193 | 7 | 13 | 12 | 13 |
| Proteins | 2 | 1091 | 32 | 303 | 19 | 160 | 4 | 7 | 3 | 5 |
| Gene | DRB1 | DRB2 | DRB3 | DRB4 | DRB5 | DRB6 | DRB7 | DRB8 | DRB9 | |
| Alleles | 1375 | 1 | 58 | 15 | 20 | 3 | 2 | 1 | 1 | |
| Proteins | 1020 | 0 | 46 | 8 | 17 | 0 | 0 | 0 | 0 |

HLA Naming can be quite confusing at first. All alleles start with "HLA", signifying they are part of the human MHC genes. The next portion (HLA-A or HLA-B) identifies which gene the allele is a modification of. The first two numbers (HLA-A*02) signifies what antigen type that particular allele is, which typically signifies the serological antigen present.[3] In other words, HLAs with the same antigen type (HLA-A*02:101 and HLA-A*02:102) will not react with each other in serological tests. The next set of digits (HLA-A*02:101) indicates what protein the allele codes for, and these are numbered sequentially in the order they are discovered. Any HLA that has a different number here produces a different protein (AKA has a nucleotide change that replaces an amino acid with another). The third set of numbers (HLA-A*02:101:01) indicates an allele variant that has a different DNA sequence but produces the same protein as the normal gene. The final set of numbers (HLA-A*02:101:01:01) is used to designate a single or multiple nucleotide polymorphism in a non-coding region of the gene. The final aspect of HLA naming is a letter (HLA-A*02:101:01:01L). There are six letters, each with a different meaning.
| Letter | Significance |
|---|---|
| N | Null allele (produces a non-functional protein) |
| L | Lower than normal cell surface expression |
| S | Soluble protein not found on cell surface |
| Q | Questionable (allele may affect normal expression) |
| C | Protein that is present in cytoplasm but not cell surface |
| A | Aberrant expression (uncertain if protein is expressed) |
Establishing the system
A person can have 2 antigen proteins per genetic-locus (one gene from each parent). When first discovered, identified antigens were clustered, creating groups in which no more than two antigens per cluster were found in a given person. Serotype group "A" consisted HL-A1, A2, A3, A9, A10, A11. Another cluster, "B", contained A7, A8, A12, A13, A14, A15. HL-A4 antigen was found to occur on lymphoid cells. Since the "HL-Antigens" no longer belonged to a single group, a new naming system was needed.
In 1968 the WHO Nomenclature Committee for Factors of the HLA System first met.[3] They established a system that divided the HLAs into HLA-A and HLA-B, A and B corresponding to a group of reactive serotypes. For example, "HL-A2" became HLA-A2, "HL-A7" became HLA-B7 and "HL-A8" became HLA-B8.
In this arrangement there were cells that were 'blank' or had new specificities, these new antigens were called "W" antigens, and as they were reassigned to new groups, for example "A" serotypes, they became Aw or Bw antigens. It was found that some antigens that behaved like A and B antigens but could be excluded based on '2-type max' exclusion. Thus a new group, "C" was created. Classification of C antigens is still ongoing, and they have retained the name Cw as many serotypes have not been developed.
The classification of the "A4" antigens was complicated. The "A4" subset evolved to become D-region antigens, which was a large cluster of genes that encoded MHC class II. Several renamings occurred. The D-region has 8 major coding loci that combine to form 3 different protein groups; DP, DQ, and DR. DRw antigens were the first to be split, a process made easy by the virtue of having an invariant alpha chain, but complicated by 4 beta chain loci (DRB1, DRB3, DRB4, and DRB5). Serotypes to DQ reacted with alpha and beta chains, or both of certain isoforms. The proper classification was greatly aided by gene sequencing and PCR. Classification and description of DP antigens is ongoing.
Genetics
Genetic complexity typifies HLA
The naming of human leukocyte antigens HLA "antigens" is deeply rooted in the discovery history of their serotypes and alleles. There is no doubt that HLA terminology can be bewildering, this terminology is a consequence of the complex genetics as well as the way these antigens were characterized.
Historical perspective is important to an understanding of how the HLA were systematized. In organ transplant the goal was to explain graft rejection for recipients, and of course, to prevent future rejection. From this perspective, the cause of rejections were found to be "antigens". In the same way bacterial antigens can cause inflammatory response, HLA antigens from the donor of the organ caused an inflammatory response when placed in a recipient. This is called allograft [allo = different, graft(medical) = transplant] rejection.
To explain rejection in a nutshell, certain immune system components are highly variable, the agents are called the Major histocompatibility (MHC) antigens. MHC antigens cause rejection of improperly matched organ transplants. The variability stems from genetics. From the perspective of human evolution, why are antigens of the MHC so variable when many other human proteins lack variability? The cause of host-versus-graft-disease may actually stem from the functions of the system.
The use of the word alloantigen actually masks the fact that HLA are infrequently autoantigens in the donor, and therefore their function is not as antigens, but something else. But the naming of these antigens is not borne out of function but the need to match organ donors with recipients.
Transplantation and transplant rejection

In the early 1960s, some physicians began more aggressive attempts at organ transplantation. Knowing little about compatibility factors, they attempted transplantation between humans and between non-humans and humans.[11] Immunosuppressive drugs worked for a time, but transplanted organs would either always fail or the patients would die from infections. Patients received skin, white blood cell or kidney donations from other donors (called allografts, meaning 'of different genetics' grafts). If these allografts were rejected, it was found that the 'rejection' response was accompanied by an antibody mediated agglutination (biology) of red blood cells (See figure).[12] The search for these cell surface antigens began. There are several processes by which antibodies can reduce function:
- Acute rejection - Antibodies could attract lymphocytes and cause them to lyse cells via the immune system's classical complement pathway
- Antibodies could bind to and alter function (e.g., flow of a fluid, or prevention of binding of ligands to receptors)
- Cytokine responses that cause systemic responses.
Different antigens can be identified
In the accompanying figure, two similar haplotypes (unknown to early clinicians) are identical, except for the one antigen in the top haplotype. The transplant may not be rejected, but if rejection does occur that allotypic protein, the alloantigen, in the donor tissue may have induced the dominant allo-reactive antibody in the recipient.
Assaying antiserum

Hemagglutination assay. In generating an immune response to an antigen, the B-cells go through a process of maturation, from surface IgM production, to serum IgM production, to maturation into a plasma cell producing IgG. Graft recipients who generate an immune response have both IgM and IgG. The IgM can be used directly in hemagglutination assays, depicted on the right. IgM has 10 antigen binding regions per molecule, allowing cross-linking of cells. An antiserum specific for HLA-A3 will then agglutinate HLA-A3 bearing red blood cells if the concentration of IgM in the antiserum is sufficiently high. Alternatively, a second antibody to the invariable (Fc) region of the IgG can be used to cross-link antibodies on different cells, causing agglutination.
Complement fixation assay. The complement fixation test was modified to assay Antiserum mediated RBC lysis.
Chromium release assay. This assay measures the release of (biological) radioactive chromium from cells as a result of killer cell activity. These cells are attracted to class I antigens that either carry foreign antigens, or are foreign to the immune system.
The role of haplotypes in identifying antigens
| Haplotype 1 | Haplotype 2 | |||||
| Example 1 | A | Cw | B | A | Cw | B |
|---|---|---|---|---|---|---|
| Donor | 1 | 7 | 8 | 3 | 7 | 7 |
| Recipient | 1 | 7 | 8 | 2 | 7 | 7 |
| Alloreactivity | 3 | |||||
| Example 2 | ||||||
| Donor | 1 | 7 | 8 | 2 | 7 | 8 |
| Recipient | 1 | 7 | 8 | 3 | 7 | 8 |
| Alloreactivity | 2 | |||||
Each person has two HLA haplotypes, a cassette of genes passed on from each parent. The haplotype frequencies in Europeans are in strong linkage disequilibrium. This means there are much higher frequencies of certain haplotypes relative to the expectation based on random sorting of gene-alleles. This aided the discovery of HLA antigens, but was unknown to the pioneering researchers.
In the tables a fortuitous transplant between two unrelated individual has resulted in an antiserum to single alloantigen. By discovering these close-but-non-identical matches, the process with somewhat related haplotypes surface antigens were identified for HLA A, and in the table below, HLA B at the time however these were all grouped together as HL-Antigens. On the left the "B" and "cw" antigens are matched (B and C are close together so if B matches then C likely also matches), but A antigens are not matched. The antisera that is produced by the recipient is most likely to be A3, but if the direction of transplant is reversed A2 is the likely alloantigen. Two of the first three alloantigens are thus readily easy to detect because of the similarity and frequency of the A2-B7 and A3-B7 haplotypes (see example 1).
| Haplotype 1 | Haplotype 2 | |||||
| Example 3 | A | Cw | B | A | Cw | B |
| Donor | 1 | 7 | 8 | 1 | 7 | 7 |
| Recipient | 1 | 7 | 8 | 1 | 7 | 8 |
| Alloreactivity | 7 | |||||
| Example 4 | ||||||
| Donor | 3 | 7 | 7 | 1 | 7 | 8 |
| Recipient | 3 | 7 | 7 | 1 | 7 | 7 |
| Alloreactivity | 8 | |||||
In these instances, the A1/A2, A2/A3, A1/A3 are matched, decreasing the probability of a rejection because many are linked to a given haplotype. Occasionally the 'recombinant' A1-Cw7-B7(rare), B7 becomes the alloantigen in a recipient with A1-Cw7-B8(common).
This linkage disequilibrium in Europeans explains why A1, A2, A3, "A7"[B7], and "A8"[B8] were identified, first. It would have taken substantially longer to identify other alleles because frequencies were lower, and haplotypes that migrated into the European population had undergone equilibration or were from multiple sources.
This is the genetic background against which scientists tried to uncover and understand the histocompatibility antigens.
A list of antigens created
In the late 1960s, scientist began reacting sera from patients with rejecting transplants to donor or 'third party' tissues. Their sera (the liquid part of the blood when blood clots) was sensitized to the cells from donors - it was alloreactive. By testing different anti-sera from recipients they were able to uncover some with unique reactivities. As a result, scientists were able to identify a few antigens. At first the first antigens were called the Hu-1 antigens[13] and tentatively tagged as gene products of the Human equivalent of the mouse histocompatibility locus (H2). In 1968, it was discovered that matching these antigens between kidney donor and recipient improved the likelihood of kidney survival in the recipient.[14] The antigen list still exists, although it has been reorganized to fit what we have since learned about genetics, refined, and greatly expanded.
Lymphocyte bearing antigens recognized

As the study of these 'rejection' sera and "allo"-antigens progressed, certain patterns in the antibody recognition were recognized. The first major observation, in 1969, was that an allotypic antibodies to "4" ("Four") was only found on lymphocytes, while most of the antigens, termed "LA", recognized most cells in the body.[15]
This group "4" antigen on lymphocytes would expand into "4a", "4b" and so on, becoming the "D" series (HLA-D (Class II) antigens) DP, DQ, and DR. This is an interesting history in itself.
The Hu-1 antigens were renamed the Human-lymphoid (HL) allo-antigens (HL-As). Allo-antigen comes from the observation that a tolerated protein in the donor becomes antigenic in the recipient. This can be compared with an autoantigen, in which a person develops antibodies to one or more of their own proteins. This also suggested the donor and recipient have a different genetic makeup for these antigens. The "LA" group thereafter was composed of HL-A1, A2, A3, A5, A6, A7, A8, A9, A10, A11, A12, A13, A14 and A15 until further divisions and renaming were necessary. Some of the antigens above, for example HL-A1, are similar to HLA-A1, as they are the same serotype. Some of the above, like A5, are not mentioned within the last few years, as they have been renamed.
During these early studies it became known that there were associations with many autoimmune diseases. And the HLA A1-B8 haplotype is linked to a very long piece of conserved chromosome 6 variant called AH8.1 haplotype. In these studies HL-A1,8 were frequently found co-linked to disease. This linkage is not necessarily a function of either gene, but a consequence of the way AH8.1 evolved.
Subclassification of lymphoid antigens
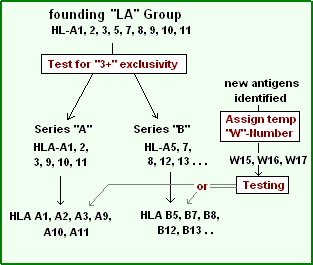
A series of tests on cultured cells revealed that, within the "LA" group, a donor tissue might have some antigens but not others. For example, an antiserum may react with patterns (on a given tissue):
- A1, A2, A7, A12
- A1, A3, A7, A8
- A1, A11, A8, A5
- A1, A8
But fail to react in the following patterns:
- A1, A2, A3, ...
- A1, A2, A11, ....
- A2, A3, A11, ....
- . . . A7, A8, A12
The HLA serotype series
Series "A"
|
If 2 members of the series (A1, 2, 3, 9, 10, 11) were typed, a reaction with a third member of the series to the donor was not observed. This 'exclusivity' identified series "A".[16] One might notice the similarities of this numeric series with the HLA-A series, as series "A" antigens are the first six members of HLA-A. Inadvertently, the scientist had discovered an antibody set that recognized only gene products from one locus, HLA-A gene the "antigens" being the gene products. The implication is that an alloreactive anti-sera can be a tool for genetic identification.
Series "B"
Not long after the series A antigens were separated from the (rapidly expanding) list of antigens, it was determined another group also could be separated along the same logical lines. This group included HL-A5, A7, A8, A12. This became the series "B". Note the similarity of Series "B" to the first few members HLA-B serotypes. The names of these antigens were necessarily changed to fit the new putative series they were assigned to. From HL-A# to HLA-B#. The problem was that the literature was using "A7" and would soon be using "B7" as shorthand for HLA-B7.
Pseudo-series "w"
Since it was now certain, by the early 1970s, that the "antigens" were encoded by different series, implicit loci, numeric lists became somewhat cumbersome. Many groups were discovering antigens. In these instances an antigen was assigned a temporary name, like "RoMa2" and after discussion, the next open numeric slot could be assigned, but not to an "A" or "B" series until proper testing had been done. To work around this problem a 'workshop' number "w#" was often assigned while testing continued to determine which series the antigen belonged to.
Series "C"
Before too long, a series "C" was uncovered. Series C has proved difficult to serotype, and the alleles in the series still carry the "w" tag signifying that status; in addition, it reminds us that Series C were not assigned names the same way as Series A and B, it has its own numeric list Cw1, Cw2, Cw3.
Serotype group expansion and refinement
By the mid 1970s, genetic research was finally beginning to make sense of the simple list of antigens, a new series "C" had been discovered and, in turn genetic research had determined the order of HLA-A, C, B and D encoding loci on the human 6p.[17] With new series came new antigens; Cw1 and 2 were quickly populated, although Cw typing lagged. Almost half of the antigens could not be resolved by serotyping in the early 90s. Currently genetics defines 18 groups.
At this point, Dw was still being used to identify DR, DQ, and DP antigens. The ability to identify new antigens far exceeded the ability to characterize those new antigens.
As technology for transplantation was deployed around the world, it became clear that these antigens were far from a complete set, and in fact hardly useful in some areas of the world (e.g., Africa, or those descended from Africans). Some serotyping antibodies proved to be poor, with broad specificities, and new serotypes were found that identified a smaller set of antigens more precisely. These broad antigen groups, like A9 and B5, were subdivided into "split" antigen groups, A23 & A24 and B51 & B52, respectively. As the HL-A serotyping developed, so did identification of new antigens.
Genetic identification
In the early 1980s, it was discovered that a restriction fragment segregates with individuals who bear the HLA-B8 serotype. By 1990, it was discovered that a single amino acid sequence difference between HLA-B44 (B*4401 versus B*4402) could result in allograft rejection. This revelation appeared to make serotyping based matching strategies problematic if many such differences existed. In the case of B44, the antigen had already been split from the B12 broad antigen group. In 1983, the cDNA sequences of HLA-A3 and Cw3[18] All three sequences compared well with mouse MHC class I antigens. The Western European HLA-B7 antigen had been sequenced (although the first sequence had errors and was replaced). In short order, many HLA class I alleles were sequenced including 2 Cw1 alleles.[19]
By 1990, the full complexity of the HLA class I antigens was beginning to be understood. At the time new serotypes were being determined, the problem with multiple alleles for each serotype was becoming apparent by nucleotide sequencing. RFLP analysis helped determine new alleles, but sequencing was more thorough. Throughout the 1990s, PCR kits, called SSP-PCR kits were developed that allowed, at least under optimal conditions, the purification of DNA, PCR and Agarose Gel identification of alleles within an 8-hour day. Alleles that could not be clearly identified by serotype and PCR could be sequenced, allowing for the refinement of new PCR kits.
Serotypes like B*4401, B*4402, B*4403, each abundant within those with B44 serotypes could be determined with unambiguous accuracy. The molecular genetics has advanced HLA technology markedly over serotyping technology, but serotyping still survives. Serotyping had identified the most similar antigens that now form the HLA subgroups. Serotyping can reveal whether an antigen coded by the relevant HLA gene is expressed. An HLA allele coding non-expressed gene is termed "Null Allele", for example: HLA-B*15:01:01:02N. The expression level can also detected by serotyping, an HLA gene coding for antigens which has low protein expression on the cell surface is termed "Low Expresser", for example: HLA-A*02:01:01:02L.
Summary
- Lymphoid "antigens" became an experimental artifact of medical techniques (i.e., of transplantation). Simply, as scientist gained familiarity with the human immune system they learned more about graft rejection, the cause was antibody production to proteins in donor tissue. The key word is allo - which means of different origin. 'Allo'typic proteins in 'allo'grafts developed immune responses in recipients. What makes these proteins different?
- From a more modern perspective, HLA gene products (i.e., antigen-presenting, cell-surface receptors) did not evolve to be transplantation antigens, nor to interfere with transplantation, organ transplantation being unknown until 1960. The HLA genes are much older. Variation in HLA major antigens is the cause of transplant rejection, but variation at HLA is under preservative selection (Called heterozygous selection or balancing selection). Variation of HLA has led to an estimate that they are at least 60 million years in age for humans (DRB1).[20] In humans, the number of HLA alleles is expanding, even with many genes, many more are still tolerable as immune presentation antigens.
The scientific problem has been to explain the natural function of a molecule, such as a self cell-surface receptor involved in immunity. It also seeks to explain how variation developed (perhaps by evolutionary pressure), and how the genetic mechanisms works (dominant, codominant, semidominant, or recessive; purifying selection or balancing selection).
References
- 1 2 3 4 5 6 7 8 9 10 11 Davis, Daniel M. The Compatibility Gene. How Our Bodies Fight Disease, Attract Others, and Define Our Selves. Oxford: Oxford UP, 2014. Print.
- ↑ Irene Park, Paul Terasaki, Origins of the first HLA specificities, Human Immunology, Volume 61, Issue 3, March 2000, Pages 185-189 doi:10.1016/S0198-8859(99)00154-8
- 1 2 3 4 5 "HLA Nomenclature @ Hla.alleles.org." HLA Nomenclature @ Hla.alleles.org. Anthony Nolan Research Institute, 10 Nov. 2013. Web. 08 Dec. 2013.
- ↑ Medawar, P. B. "A second study of the behaviour and fate of skin homografts in rabbits: a report to the War Wounds Committee of the Medical Research Council. Journal of Anatomy 1945; 79, 157-76
- ↑ Billingham, R.E., Brent, L. and Medawar, P.B. "Quantitative studies on tissue transplantation immunity. iii. Actively acquired tolerance. Philosophical Transactions of the Royal Society of London B Biological Sciences 1956; 239, 357-414
- ↑ Davis, Daniel M. The Compatibility Gene. How Our Bodies Fight Disease, Attract Others, and Define Our Selves. Oxford: Oxford UP, 2014. Print. pg 34
- ↑ Madura, Florian, Pierre J. Rizkallah, Kim M. Miles, Christopher J. Holland, Anna M. Bulek, Anna Fuller, Andrea J. A. Schauenburg, John J. Miles, Nathaniel Liddy, Malkit Sami, Yi Li, Moushumi Hossain, Brian M. Baker, Bent K. Jakobsen, Andrew K. Sewell, and David K. Cole. "T-cell Receptor Specificity Maintained by Altered Thermodynamics." Journal of Biological Chemistry 288 (June 2013): 18766-18775.
- ↑ Doherty, P.C. and Zinkernagel, R.M. A biological role for the major histocompatibility antigens. Lancet I, 1406-9 (1975).
- ↑ Alberts, Bruce. Essential Cell Biology. New York: Garland Science, 2009. Print.
- 1 2 3 "Statistics." IPD- IMGT/HLA. European Molecular Biology Lab, 2013. Web. 13 Dec. 2013.
- ↑ Reemtsma K, Mccracken BH, Schlegel JU, Pearl M (1964). "Heterotransplantation of the kidney: two clinical experiences". Science. 143 (3607): 700–2. doi:10.1126/science.143.3607.700. PMID 14081245.
- ↑ Rapaport FT, Kano K, Milgrom F (1968). "Heterophile antibodies in human transplantation". J. Clin. Invest. 47 (3): 633–42. doi:10.1172/JCI105759. PMC 297209
 . PMID 4866325.
. PMID 4866325. - ↑ Bach FH, Amos DB (1967). "Hu-1: Major histocompatibility locus in man". Science. 156 (3781): 1506–8. doi:10.1126/science.156.3781.1506. PMID 4887739.
- ↑ Patel R, Mickey MR, Terasaki PI (1968). "Serotyping for homotransplantation. XVI. Analysis of kidney transplants from unrelated donors". N. Engl. J. Med. 279 (10): 501–6. doi:10.1056/NEJM196809052791001. PMID 4876470.
- ↑ Mann DL, Rogentine GN, Fahey JL, Nathenson SG (1969). "Molecular heterogeneity of human lymphoid (HL-A) alloantigens". Science. 163 (3874): 1460–2. doi:10.1126/science.163.3874.1460. PMID 5773111.
- ↑ Bach ML, Bach FH. (1970) The genetics of histocompatibility. Hosp. Practice 5(8): 33-44
- ↑ Yunis EJ, Dupont B, Hansen J (1976). "Immunogenetic aspects of allotransplantation". Adv. Exp. Med. Biol. 73 Pt B: 231–51. doi:10.1007/978-1-4684-3300-5_20. PMID 136874.
- ↑ Strachan T, Sodoyer R, Damotte M, Jordan BR (1984). "Complete nucleotide sequence of a functional class I HLA gene, HLA-A3: implications for the evolution of HLA genes". EMBO J. 3 (4): 887–94. PMC 557443. PMID 6609814.
- ↑ Parham P, Lomen CE, Lawlor DA, et al. (1988). "Nature of polymorphism in HLA-A, -B, and -C molecules". Proc. Natl. Acad. Sci. U.S.A. 85 (11): 4005–9. doi:10.1073/pnas.85.11.4005. PMC 280349. PMID 3375250.
- ↑ Ayala FJ (1995). "The myth of Eve: molecular biology and human origins". Science. 270 (5244): 1930–6. doi:10.1126/science.270.5244.1930. PMID 8533083.