Determination of equilibrium constants
Equilibrium constants are determined in order to quantify chemical equilibria. When an equilibrium constant K is expressed as a concentration quotient,
![{\displaystyle K={\frac {\mathrm {[S]} ^{\sigma }\mathrm {[T]} ^{\tau }\cdots }{\mathrm {[A]} ^{\alpha }\mathrm {[B]} ^{\beta }\cdots }}}](../I/m/7fbf3623d6284c219011f318e2197779ab194931.svg)
it is implied that the activity quotient is constant. For this assumption to be valid, equilibrium constants must be determined in a medium of relatively high ionic strength. Where this is not possible, consideration should be given to possible activity variation.
The equilibrium expression above is a function of the concentrations [A], [B] etc. of the chemical species in equilibrium. The equilibrium constant value can be determined if any one of these concentrations can be measured. The general procedure is that the concentration in question is measured for a series of solutions with known analytical concentrations of the reactants. Typically, a titration is performed with one or more reactants in the titration vessel and one or more reactants in the burette. Knowing the analytical concentrations of reactants initially in the reaction vessel and in the burette, all analytical concentrations can be derived as a function of the volume (or mass) of titrant added.
The equilibrium constants may be derived by best-fitting of the experimental data with a chemical model of the equilibrium system.
Experimental methods
There are four main experimental methods. For less commonly used methods, see Rossotti and Rossotti.[1]
Potentiometric measurements
A free concentration [A] or activity {A} of a species A is measured by means of an ion selective electrode such as the glass electrode. If the electrode is calibrated using activity standards it is assumed that the Nernst equation applies in the form

where E0 is the standard electrode potential. When buffer solutions of known pH are used for calibration the meter reading will be a pH.

At 298 K, 1 pH unit is approximately equal to 59 mV.[2]
When the electrode is calibrated with solutions of known concentration, by means of a strong acid–strong base titration, for example, a modified Nernst equation is assumed.
![{\displaystyle E=E^{0}+s\log _{10}\mathrm {[A]} }](../I/m/c404b52bc56775c11adfd79fab6c73166abc0193.svg)
where s is an empirical slope factor. A solution of known hydrogen ion concentration may be prepared by standardization of a strong acid against borax. Constant-boiling hydrochloric acid may also be used as a primary standard for hydrogen ion concentration.
Spectrophotometric measurements
Absorbance
It is assumed that the Beer–Lambert law applies.

where l is the optical path length, ε is a molar absorbance at unit path length and c is a concentration. More than one of the species may contribute to the absorbance. In principle absorbance may be measured at one wavelength only, but in present-day practice it is common to record complete spectra.
Fluorescence (luminescence) intensity
It is assumed that the scattered light intensity is a linear function of species’ concentrations.

where φ is a proportionality constant.
NMR chemical shift measurements
Chemical exchange is assumed to be rapid on the NMR time-scale. An individual chemical shift δ is the mole-fraction-weighted average of the shifts δ of nuclei in contributing species.

Example: the pKa of the hydroxyl group in citric acid has been determined from 13C chemical shift data to be 14.4. Neither potentiometry nor UV–visible spectrophotometry could be used for this determination.[3]
Calorimetric measurements
Simultaneous measurement of K and ΔH for 1:1 adducts is routinely carried out using isothermal titration calorimetry. Extension to more complex systems is limited by the availability of suitable software.
Range and limitations
- Potentiometry
- The most widely used electrode is the glass electrode, which is selective for the hydrogen ion. This is suitable for all acid–base equilibria. log10 β values between about 2 and 11 can be measured directly by potentiometric titration using a glass electrode. This enormous range is possible because of the logarithmic response of the electrode. The limitations arise because the Nernst equation breaks down at very low or very high pH. The range can be extended by using the competition method. An example of the application of this method can be found in palladium(II) cyanide.
- Absorbance and luminescence
- An upper limit on log10 β of 4 is usually quoted, corresponding to the precision of the measurements, but it also depends on how intense the effect is. Spectra of contributing species should be clearly distinct from each other
- NMR
- Limited precision of chemical shift measurements also puts an upper limit of about 4 on log10 β. Limited to diamagnetic systems. 1H NMR cannot be used with solutions of compounds in 1H2O.
- Calorimetry
- Insufficient evidence is currently available.
Computational methods
It is assumed that the collected experimental data comprise a set of data points. At each ith data point, the analytical concentrations of the reactants, TA(i), TB(i) etc. are known along with a measured quantity, yi, that depends on one or more of these analytical concentrations. A general computational procedure has four main components:
- Definition of a chemical model of the equilibria
- Calculation of the concentrations of all the chemical species in each solution
- Refinement of the equilibrium constants
- Model selection
The chemical model
The chemical model consists of a set of chemical species present in solution, both the reactants added to the reaction mixture and the complex species formed from them. Denoting the reactants by A, B..., each complex species is specified by the stoichiometric coefficients that relate the particular combination of reactants forming them.
- :

![{\displaystyle \beta _{pq\cdots }={\frac {[{\ce {A}}_{p}{\ce {B}}_{q}\cdots ]}{[{\ce {A}}]^{p}[{\ce {B}}]^{q}\cdots }}}](../I/m/41ddfaa95c2aab1288ee0ef5881d7829e498e933.svg)
When using general-purpose computer programs, it is usual to use cumulative association constants, as shown above. Electrical charges are not shown in general expressions such as this and are often omitted from specific expressions, for simplicity of notation. In fact, electrical charges have no bearing on the equilibrium processes other that there being a requirement for overall electrical neutrality in all systems.
With aqueous solutions the concentrations of proton (hydronium ion) and hydroxide ion are constrained by the self-dissociation of water.
- :

![{\displaystyle K_{\mathrm {W} }^{'}={\frac {\ce {[H+][OH^{-}]}}{\ce {[H2O]}}}}](../I/m/a3dd322a0f1da045b23b286b517be09714a8eff9.svg)
With dilute solutions the concentration of water is assumed constant, so the equilibrium expression is written in the form of the ionic product of water.
![{\displaystyle K_{\mathrm {W} }={\ce {[H+][OH^{-}]}}\,}](../I/m/20c205436413230cb49ee0120e9e09d08ceb57f6.svg)
When both H+ and OH− must be considered as reactants, one of them is eliminated from the model by specifying that its concentration be derived from the concentration of the other. Usually the concentration of the hydroxide ion is given by
![{\displaystyle {\ce {[OH^{-}]}}={\frac {K_{\mathrm {W} }}{[{\ce {H+}}]}}\,}](../I/m/64fbcd96d27d0edc7e75b64f0ae20404d8d0a44a.svg)
In this case the equilibrium constant for the formation of hydroxide has the stoichiometric coefficients −1 in regard to the proton and zero for the other reactants. This has important implications for all protonation equilibria in aqueous solution and for hydrolysis constants in particular.
It is quite usual to omit from the model those species whose concentrations are considered negligible. For example, it is usually assumed then there is no interaction between the reactants and/or complexes and the electrolyte used to maintain constant ionic strength or the buffer used to maintain constant pH. These assumptions may or may not be justified. Also, it is implicitly assumed that there are no other complex species present. When complexes are wrongly ignored a systematic error is introduced into the calculations.
Equilibrium constant values are usually estimated initially by reference to data sources.
Speciation calculations
A speciation calculation is one in which concentrations of all the species in an equilibrium system are calculated, knowing the analytical concentrations, TA, TB etc. of the reactants A, B etc. This means solving a set of nonlinear equations of mass-balance
![{\displaystyle {\begin{aligned}T_{\ce {A}}&=[{\ce {A}}]+\sum p\beta _{pq\cdots }[{\ce {A}}]^{p}[{\ce {B}}]^{q}\cdots \\[8pt]T_{\ce {B}}&=[{\ce {B}}]+\sum q\beta _{pq\cdots }[{\ce {A}}]^{p}[{\ce {B}}]^{q}\cdots \\&{}\ \ \vdots \end{aligned}}}](../I/m/e711e6094b26d2a12c8e72e6c684ebfc4e85cbe0.svg)
for the free concentrations [A], [B] etc. The concentrations of the complexes are derived from the free concentrations via the chemical model. Some authors[4][5] include the free reactant terms in the sums by declaring identity (unit) β constants for which the stoichiometric coefficients are 1 for the reactant concerned and zero for all other reactants:
![{\displaystyle {\ce {[A]=\beta _{10\ldots }[A],[B]=\beta _{01\ldots }[B]\ldots \,}}}](../I/m/a2d5e5fbe1501efe866b8aa6d6080443e2eb0c32.svg)

In this manner, all chemical species, including the free reactants, are treated in the same way, having been formed from the combination of reactants that is specified by the stoichiometric coefficients. The mass-balance equations assume the simpler form.
![{\displaystyle {\begin{aligned}T_{\ce {A}}&=\sum p\beta _{pq\cdots }[{\ce {A}}]^{p}[{\ce {B}}]^{q}\cdots \\[8pt]T_{\ce {B}}&=\sum q\beta _{pq\cdots }[{\ce {A}}]^{p}[{\ce {B}}]^{q}\cdots \\&{}\ \ \vdots \end{aligned}}}](../I/m/fa3c1ae46892f52bf6e7774eac685b753b5dc9f9.svg)
In a titration system the analytical concentrations of the reactants at each titration point are obtained from the initial conditions, the burette concentrations and volumes. The analytical (total) concentration of a reactant R at the ith titration point is given by
![{\displaystyle T_{\ce {R}}={\frac {{\ce {R}}_{0}+v_{i}{\ce {[R]}}}{v_{0}+v_{i}}}}](../I/m/8016e8729734f7daca35e37aa7f1a3f648cf429f.svg)
where R0 is the initial amount of R in the titration vessel, v0 is the initial volume, [R] is the concentration of R in the burette and vi is the volume added. The burette concentration of a reactant not present in the burette is taken to be zero.
In general, solving these nonlinear equations presents a formidable challenge because of the huge range over which the free concentrations may vary. At the beginning, values for the free concentrations must be estimated. Then, these values are refined, usually by means of Newton–Raphson iterations. The logarithms of the free concentrations may be refined rather than the free concentrations themselves. Refinement of the logarithms of the free concentrations has the added advantage of automatically imposing a non-negativity constraint on the free concentrations. Once the free reactant concentrations have been calculated, the concentrations of the complexes are derived from them and the equilibrium constants.
Note that the free reactant concentrations can be regarded as implicit parameters in the equilibrium constant refinement process. In that context the values of the free concentrations are constrained by forcing the conditions of mass-balance to apply at all stages of the process.
Equilibrium constant refinement
The objective of the refinement process is to find equilibrium constant values that give the best fit to the experimental data. This is usually achieved by minimising an objective function, U, by the method of non-linear least-squares. First the residuals are defined as

Then the most general objective function is given by

The matrix of weights, W, should be, ideally, the inverse of the variance-covariance matrix of the observations. It is rare for this to be known. However, when it is, the expectation value of U is one, which means that the data are fitted within experimental error. Most often only the diagonal elements are known, in which case the objective function simplifies to

with Wij = 0 when j ≠ i. Unit weights, Wii = 1, are often used but, in that case, the expectation value of U is the root mean square of the experimental errors.
The minimization may be performed using the Gauss–Newton method. Firstly the objective function is linearised by approximating it as a first-order Taylor series expansion about an initial parameter set, p.

The increments δpi are added to the corresponding initial parameters such that U is less than U0. At the minimum the derivatives ∂U/∂pi, which are simply related to the elements of the Jacobian matrix, J

where pk is the kth parameter of the refinement, are equal to zero. One or more equilibrium constants may be parameters of the refinement. However, the measured quantities (see above) represented by y are not expressed in terms of the equilibrium constants, but in terms of the species concentrations, which are implicit functions of these parameters. Therefore, the Jacobian elements must be obtained using implicit differentiation.
The parameter increments δp are calculated by solving the normal equations, derived from the conditions that ∂U/∂p = 0 at the minimum.

The increments δp are added iteratively to the parameters

where n is an iteration number. The species concentrations and ycalc values are recalculated at every data point. The iterations are continued until no significant reduction in U is achieved, that is, until a convergence criterion is satisfied. If, however, the updated parameters do not result in a decrease of the objective function, that is, if divergence occurs, the increment calculation must be modified. The simplest modification is to use a fraction, f, of calculated increment, so-called shift-cutting.

In this case, the direction of the shift vector, δp, is unchanged. With the more powerful Levenberg–Marquardt algorithm, on the other hand, the shift vector is rotated towards the direction of steepest descent, by modifying the normal equations,

where λ is the Marquardt parameter and I is an identity matrix. Other methods of handling divergence have been proposed.[5]
A particular issue arises with NMR and spectrophotometric data. For the latter, the observed quantity is absorbance, A, and the Beer–Lambert law can be written as

It can be seen that absorbance, A, is a linear function of the molar absorbptivities, ε, at the path length used. In matrix notation

There are two approaches to the calculation of the unknown molar absorptivities
- The ε values are considered parameters of the minimization and the Jacobian is constructed on that basis. However, the ε values themselves are calculated at each step of the refinement by linear least-squares:
- using the refined values of the equilibrium constants to obtain the speciation. The matrix
- is an example of a pseudo-inverse.


- The Beer–Lambert law is written as
- Golub and Pereyra[6] showed how the pseudo-inverse can be differentiated so that parameter increments for both molar absorptivities and equilibrium constants can be calculated by solving the normal equations.

Parameter errors and correlation
In the region close to the minimum of the objective function, U, the system approximates to a linear least-squares system, for which

Therefore, the parameter values are (approximately) linear combinations of the observed data values and the errors on the parameters, p, can be obtained by error propagation from the observations, yobs, using the linear formula. Let the variance-covariance matrix for the observations be denoted by Σy and that of the parameters by Σp. Then,

When W = (Σy)−1, this simplifies to

In most cases the errors on the observations are un-correlated, so that Σy is diagonal. If so, each weight should be the reciprocal of the variance of the corresponding observation. For example, in a potentiometric titration, the weight at a titration point, k, can be given by

where σE is the error in electrode potential or pH, (∂E/∂v)
k is the slope of the titration curve and σv is the error on added volume.
When unit weights are used (W = I, p = (JTJ)−1JTy) it is implied that the experimental errors are uncorrelated and all equal: Σy = σ2I, where σ2 is known as the variance of an observation of unit weight, and I is an identity matrix. In this case σ2 is approximated by

where U is the minimum value of the objective function and nd and np are the number of data and parameters, respectively.

In all cases, the variance of the parameter pi is given by Σp
ii and the covariance between parameters pi and pj is given by Σp
ij. Standard deviation is the square root of variance. These error estimates reflect only random errors in the measurements. The true uncertainty in the parameters is larger due to the presence of systematic errors—which, by definition, cannot be quantified.
Note that even though the observations may be uncorrelated, the parameters are always correlated.
Derived constants
When cumulative constants have been refined it is often useful to derive stepwise constants from them. The general procedure is to write down the defining expressions for all the constants involved and then to equate concentrations. For example, suppose that one wishes to derive the pKa for removing one proton from a tribasic acid, LH3, such as citric acid.
![{\displaystyle {\begin{array}{ll}{\ce {{L^{3-}}+{2H+}<=>LH2^{-}}};&{\ce {[LH2^{-}]=\beta _{12}[L^{3-}][H+]^{2}}}\\{\ce {{L^{3-}}+{3H+}<=>LH3}};&{\ce {[LH3]=\beta _{13}[L^{3-}][H+]^{3}}}\end{array}}}](../I/m/b36b8e642a26eb866580aebc57c9fd5fc987a7b2.svg)
The stepwise association constant for formation of LH3 is given by
![{\displaystyle {\ce {{LH2^{-}}+{H+}<=>LH_{3}}};\quad \ [{\ce {LH3}}]=K{\ce {[LH2^{-}][H+]}}}](../I/m/3b601db6585887730f5fd5fbd9722fc6c0081b9d.svg)
Substitute the expressions for the concentrations of LH3 and LH−
2 into this equation
![{\displaystyle \beta _{13}{\ce {[L^{3-}][H+]^{3}}}=K\beta _{12}{\ce {[L^{3-}][H+]^{2}[H+]}}\,}](../I/m/a6e24eb1694319123d0d0acb89309b73d96e82b1.svg)
whence

and since pKa = −log10 1/K its value is given by

When calculating the error on the stepwise constant, the fact that the cumulative constants are correlated must accounted for. By error propagation

and

Model selection
Once a refinement has been completed the results should be checked to verify that the chosen model is acceptable. generally speaking, a model is acceptable when the data are fitted within experimental error, but there is no single criterion to use to make the judgement. The following should be considered.
The objective function
When the weights have been correctly derived from estimates of experimental error, the expectation value of U/nd − np is 1.[7] It is therefore very useful to estimate experimental errors and derive some reasonable weights from them as this is an absolute indicator of the goodness of fit.
When unit weights are used, it is implied that all observations have the same variance. U/nd − np is expected to be equal to that variance.
Parameter errors
One would want the errors on the stability constants to be roughly commensurate with experimental error. For example, with pH titration data, if pH is measured to 2 decimal places, the errors of log10 β should not be much larger than 0.01. In exploratory work where the nature of the species present is not known in advance, several different chemical models may be tested and compared. There will be models where the uncertainties in the best estimate of an equilibrium constant may be somewhat or even significantly larger than σpH, especially with those constants governing the formation of comparatively minor species, but the decision as to how large is acceptable remains subjective. The decision process as to whether or not to include comparatively uncertain equilibria in a model, and for the comparison of competing models in general, can be made objective and has been outlined by Hamilton.[7]
Distribution of residuals
At the minimum in U the system can be approximated to a linear one, the residuals in the case of unit weights are related to the observations by

The symmetric, idempotent matrix J(JTT)−1J is known in the statistics literature as the hat matrix, H. Thus,

and

where I is an identity matrix and Mr and My are the variance-covariance matrices of the residuals and observations, respectively. This shows that even though the observations may be uncorrelated, the residuals are always correlated.
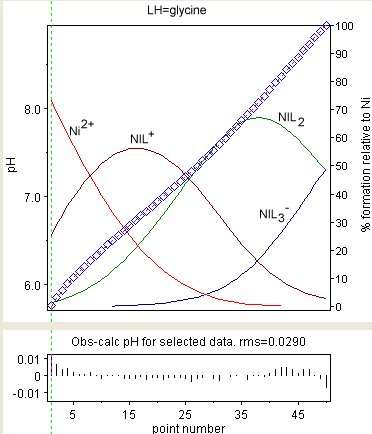
The diagram at the right shows the result of a refinement of the stability constants of Ni(Gly)+, Ni(Gly)2 and Ni(Gly)−
3 (where GlyH = glycine). The observed values are shown a blue diamonds and the species concentrations, as a percentage of the total nickel, are superimposed. The residuals are shown in the lower box. The presence of correlation is evident in the way sequences all have the same sign. Correlation notwithstanding, the magnitudes of the residuals show some randomness. Individual residuals are mostly commensurate with experimental error (about 0.002 in pH). This is about as good as it gets.
Physical constraints
Some physical constraints are usually incorporated in the calculations. For example, all the concentrations of free reactants and species must have positive values and association constants must have positive values.
With spectrophotometric data the molar absorptivity (or emissivity) values should all be positive. Most computer programs do not impose this constraint on the calculations.
Other models
If the model is not acceptable, a variety of other models should be examined to find one that best fits the experimental data, within experimental error. The main difficulty is with the so-called minor species. These are species whose concentration is so low that the effect on the measured quantity is at or below the level of error in the experimental measurement. The constant for a minor species may prove impossible to determine if there is no means to increase the concentration of the species. .
Implementations
Some simple systems are amenable to spreadsheet calculations.[8] A large number of computer programs for equilibrium constant calculation have been published. See [9] for a bibliography. The most frequently used programs are:
- Potentiometric data: Hyperquad, BEST[10] PSEQUAD[11]
- Spectrophotometric data:HypSpec, SQUAD,[11] Specfit,[12] ReactLab EQUILIBRIA.
- NMR data HypNMR, EQNMR
References
- ↑ Rossotti, F. J. C.; Rossotti, H. (1961). The Determination of Stability Constants. McGraw-Hill.
- ↑ "Definitions of pH scales, standard reference values, measurement of pH, and related terminology" (PDF). Pure Appl. Chem. 57: 531–542. 1985. doi:10.1351/pac198557030531.
- ↑ Silva, Andre M. N.; Kong, Xiaole; Hider, Robert C. (2009). "Determination of the pKa value of the hydroxyl group in the α-hydroxycarboxylates citrate, malate and lactate by 13C NMR: implications for metal coordination in biological systems". Biometals. 22 (5): 771–778. doi:10.1007/s10534-009-9224-5.
- ↑ Motekaitis, R. J.; Martell, A. E. (1982). Can. J. Chem. 60: 2403–2409. doi:10.1139/v82-347. Missing or empty
|title=(help) - 1 2 Potvin, P. G. (1990). Can. J. Chem. 68: 2198–2207. doi:10.1139/v90-337. Missing or empty
|title=(help) - ↑ Golub, G. H.; Pereyra, V. (1973). SIAM J. Numer. Anal. 2: 413–432. doi:10.1137/0710036. Missing or empty
|title=(help) - 1 2 Hamilton, W. C. (1964). Statistics in Physical Science. New York, NY: Ronald Press.
- ↑ Billo, E. Joseph (2011). Excel for Chemists: A Comprehensive Guide (3rd ed.). Wiley-VCH. ISBN 978-0-470-38123-6.
- ↑ Gans, P.; Sabatini, A.; Vacca, A. (1996). "Talanta". 43: 1739–1753. doi:10.1016/0039-9140(96)01958-3.
- ↑ Martell, A. E.; Motekaitis, R. J. (1992). The Determination and Use of Stability Constants. Wiley-VCH. ISBN 0471188174.
- 1 2 Leggett, D. J., ed. (1985). Computational Methods for the Determination of Formation Constants. Plenum Press. ISBN 978-0-306-41957-7.
- ↑ Gampp, H.; Maeder, M.; Mayer, C. J.; Zuberbühler, A. (1985). Talanta. 32 (95): 257. doi:10.1016/0039-9140(85)80035-7. Missing or empty
|title=(help)