dbSNP
| Content | |
|---|---|
| Description | Single Nucleotide Polymorphism Database |
| Organisms | all |
| Contact | |
| Research center | National Center for Biotechnology Information |
| Primary citation | PMID 21097890 |
| Release date | 1998 |
| Access | |
| Data format | ASN.1, Fasta, XML |
| Website |
www |
| Download URL | ftp://ftp.ncbi.nih.gov/snp/ |
| Web service URL |
EUtils SOAP |
The Single Nucleotide Polymorphism Database[1] (dbSNP) is a free public archive for genetic variation within and across different species developed and hosted by the National Center for Biotechnology Information (NCBI) in collaboration with the National Human Genome Research Institute (NHGRI). Although the name of the database implies a collection of one class of polymorphisms only (i.e., single nucleotide polymorphisms (SNPs)), it in fact contains a range of molecular variation: (1) SNPs, (2) short deletion and insertion polymorphisms (indels/DIPs), (3) microsatellite markers or short tandem repeats (STRs), (4) multinucleotide polymorphisms (MNPs), (5) heterozygous sequences, and (6) named variants.[2] The dbSNP accepts apparently neutral polymorphisms, polymorphisms corresponding to known phenotypes, and regions of no variation. It was created in September 1998 to supplement GenBank, NCBI’s collection of publicly available nucleic acid and protein sequences.[2]
As of build 131 (available February 2010), dbSNP had amassed over 184 million submissions representing more than 64 million distinct variants for 55 organisms, including Homo sapiens, Mus musculus, Oryza sativa, and many other species. A full list of organisms and the number of submissions for each can be found at: http://www.ncbi.nlm.nih.gov/SNP/snp_summary.cgi
Purpose
dbSNP is an online resource implemented to aid biology researchers. Its goal is to act as a single database that contains all identified genetic variation, which can be used to investigate a wide variety of genetically based natural phenomenon. Specifically, access to the molecular variation cataloged within dbSNP aids basic research such as physical mapping, population genetics, investigations into evolutionary relationships, as well as being able to quickly and easily quantify the amount of variation at a given site of interest. In addition, dbSNP guides applied research in pharmacogenomics and the association of genetic variation with phenotypic traits.[3] According to the NCBI website, “The long-term investment in such novel and exciting research [dbSNP] promises not only to advance human biology but to revolutionise the practice of modern medicine.”
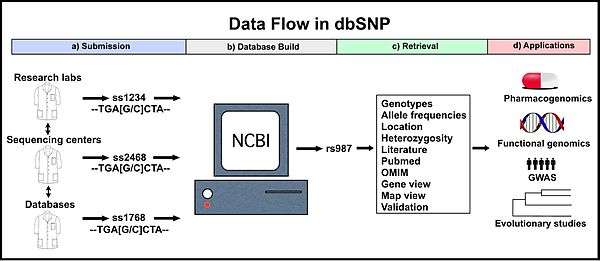
Submission
1. Source
dbSNP accepts submissions for any organism from a wide variety of sources including individual research laboratories, collaborative polymorphism discovery efforts, large scale genome sequencing centers, other SNP databases (e.g. the SNP consortium, HapMap, etc.), and private businesses.[4]
2. Types of records
Every submitted variation receives a submitted SNP ID number (“ss#”).[4] This accession number is a stable and unique identifier for that submission. Unique submitted SNP records also receive a reference SNP ID number (“rs#”; "refSNP cluster"). However, more than one record of a variation will likely be submitted to dbSNP, especially for clinically relevant variations. To accommodate this, dbSNP routinely assembles identical submitted SNP records into a single reference SNP record, which is also a unique and stable identifier (see below).[3]
3. How to submit
To submit variations to dbSNP, one must first acquire a submitter handle, which identifies the laboratory responsible for the submission.[3] Next, the author is required to complete a submission file containing the relevant information and data. Submitted records must contain the ten essential pieces of information listed in the following table.[3] Other information required for submissions includes contact information, publication information (title, journal, authors, year), molecule type (genomic DNA, cDNA, mitochondrial DNA, chloroplast DNA), and organism.[3] A sample submission sheet can be found at: (http://www.ncbi.nlm.nih.gov/SNP/get_html.cgi?whichHtml=how_to_submit#SECTION_TYPES)
| Element | Explanation |
|---|---|
| Flanking DNA | Variations from assays must have 25 bp of flanking sequence on either side of the polymorphism and must be 100 bp overall. |
| Alleles | Alleles must be defined using A, G, C, or T nomenclature; IUPAC nomenclature will only be accepted in flanking regions. |
| Method | A description of how the variation was detected (e.g. DNA sequencing) or how the allele frequencies were calculated. A table of method classes is provided. |
| Population | A description of the initial group from which the variation was found or from which the allele frequency was calculated. A table of population classes is provided. |
| Sample size | The number of chromosomes used to find the variation and the number of chromosomes used to calculate allele frequencies. |
| Population-specific allele frequency | The allele frequency of the surveyed population. |
| Population-specific genotype frequency | The genotype frequency of the surveyed population. |
| Population-specific heterozygosity | The proportion of individuals who are heterozygous for the variation. |
| Individual genotypes | The genotype of individuals from the study. |
| Validation information | The validation status lists the categories of evidence supporting the variation. |
Release
New information obtained by dbSNP becomes available to the public periodically in a series of “builds” (i.e. revisions and releases of data).[3] There is no schedule for releasing new builds; instead, builds are usually released when a new genome build becomes available, assuming that the genome has some cataloged variation associated with it.[5] This occurs approximately every 1–2 months. Genome sequences often contain errors so reference SNPs (“refSNP”) from previous builds, as well as new submitted SNPs, are re-mapped to the newly available genome sequence through multiple cycles of BLAST[6] and MegaBLAST. Multiple submitted SNPs, if mapping to the same location, are clustered into one refSNP cluster and are assigned a reference SNP ID number. However, if two refSNP cluster records are found to map to the same location (i.e. are identical), then dbSNP will also merge those records together. In this case, the smallest refSNP number ID (i.e. the earliest record) would now represent both records, and the larger refSNP number IDs would become obsolete. These obsolete refSNP number IDs and are not used again for new records. When a merger of two refSNP records occurs, the change is tracked, and the former refSNP number IDs can still be used as a search query. This process of merging identical records together reduces redundancy within dbSNP.[5]
There are two exceptions to the above merging criteria. First, variation of different classes (e.g. a SNP and a DIP) are not merged. Secondly, clinically important refSNPs that have been cited in the literature are termed “precious”; a merger that would eliminate such a refSNP is never performed, since it could later cause confusion.[5]
Retrieval
1. How to
The dbSNP can be searched using the Entrez SNP search tool (found at http://www.ncbi.nlm.nih.gov/projects/SNP/). A variety of queries can be used for searching: an ss number ID, a refSNP number ID, a gene name, an experimental method, a population class, a population detail, a publication, a marker, an allele, a chromosome, a base position, a heterozygosity range, a build number, or a strain.[5][7] In addition, many results can be retrieved simultaneously using batch queries.[5] Searches return refSNP number IDs that match the query term and a summary of the available information for that refSNP cluster.
2. Tools/Data
The information available for a refSNP cluster includes the basic information from each of the individual submissions (see “Submission”) as well as information available from combining the data from multiple submissions (e.g. heterozygosity, genotype frequencies). Many tools are available to examine a refSNP cluster in greater depth. Map view shows the position of the variation in the genome and other nearby variations. Another tool, gene view reports the location of the variation within a gene (if it is in a gene), the old and new codon, the amino acids encoded by both, and whether the change is synonymous or non-synonymous. Sequence viewer shows the position of the variant in relation to introns, exons, and other distant and close variants. 3D structure mapping, which shows 3D images of the encoded protein is also available.
The dbSNP is also linked to many other NCBI resources including the nucleotide, protein, gene, taxonomy and structure databases, as well as PubMed, UniSTS, PMC, OMIM, and UniGene.
3. Validation status
The validation status list the categories of evidence that support a variant. These include: (1) multiple independent submissions; (2) frequency or genotype data; (3) submitter confirmation; (4) observation of all alleles in at least two chromosomes; (5) genotyped by HapMap; and (6) sequenced in the 1000 Genomes Project.[5]
Problems
The quality of the data found on dbSNP has been questioned by many research groups [8][9][10][11][12][13] , which suspect high false positive rates due to genotyping and base-calling errors. These mistakes can easily be entered into dbSNP if the submitter uses (1) uncritical bioinformatic alignments of highly similar but distinct DNA sequences, and/or (2) PCRs with primers that cannot discriminate between similar but distinct DNA sequences.[8] Mitchell et al. (2004) [9] reviewed four studies [10][11][12][13] and concluded that dbSNP has a false positive rate between 15-17% for SNPs, and also that the minor allele frequency is greater than 10% for approximately 80% of the SNPs that are not false positives. Similarly, Musemeci et al. (2010)[8] states that as many as 8.32% of the biallelic coding SNPs in dbSNP are artifacts of highly similar DNA sequences (i.e. paralogous genes) and refer to these entries as single nucleotide differences (SNDs). The high error rates in dbSNP may not be surprising: of the 23.7 million refSNP entries for humans, only 14.5 million have been validated, leaving the remaining 9.2 million as candidate SNPs. However, according to Musemeci et al. (2010),[8] even the validation code provided in the refSNP record is only partially useful: only HapMap validation reduced the number of SNDs (3% vs 8%), but only accepting this method removes more than half of the real SNPs in the dbSNP. These authors also note that one source of submissions from the Lee group are plagued with errors: 20% of these submissions are SNDs (vs. 8% for submissions). However, as the authors note, ignoring all of these submissions would remove many real SNPs.
Errors in the dbSNP can hamper candidate gene association studies[14] and haplotype-based investigations.[15] Errors may also increase false conclusions in association studies:[8] increasing the number of SNPs that are tested by testing false SNPs requires more hypothesis tests. However, these false SNPs cannot actually be associated with traits, so the alpha level is decreased more than is necessary for a rigorous test if only the true SNPs were tested and the false negative rate will increase. Musemeci et al. (2010)[8] suggested that authors of negative association studies inspect their previous studies for false SNPs (SNDs), which could be removed from analysis.
How to cite data from dbSNP
Individual sequences can be referred to by their refSNP cluster ID numbers (e.g. rs206437). dbSNP should be referenced using the 2001 Sherry et al. paper: Sherry, S.T., Ward, M.H., Kholodov, M., Baker, J., Phan, L., Smigielski, E.M., Sirotkin, K. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acid Research, 29: 308-311.[4]
See also
References
- ↑ Wheeler DL, Barrett T, Benson DA, et al. (January 2007). "Database resources of the National Center for Biotechnology Information". Nucleic Acids Res. 35 (Database issue): D5–12. doi:10.1093/nar/gkl1031. PMC 1781113
 . PMID 17170002.
. PMID 17170002. - 1 2 Sherry ST, Ward M; Sirotkin, K. (1999). "dbSNP - database for single nucleotide polymorphisms and other classes of minor genetic variation". Genome Research. 9 (8): 677–679. doi:10.1101/gr.9.8.677. PMID 10447503.
- 1 2 3 4 5 6 Kitts A; Sherry S, (2009). "The single nucleotide polymorphism database (dbSNP) of nucleotide sequence variation".
- 1 2 3 Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K, et al. (2001). "dbSNP: the NCBI database of genetic variation". Nucleic Acids Res. 29 (1): 308–311. doi:10.1093/nar/29.1.308. PMC 29783. PMID 11125122.
- 1 2 3 4 5 6 NCBI (2010). "The single nucleotide polymorphism database (dbSNP) frequently asked questions".
- ↑ Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, et al. (1990). "Basic local alignment search tool". Journal of Molecular Biology. 215 (3): 403–410. doi:10.1016/S0022-2836(05)80360-2. PMID 2231712.
- ↑ Phillips, C (2007). "Online resources for SNP analysis: A review and route map". Molecular Biotechnology. 35 (1): 65–97. doi:10.1385/MB:35:1:65. PMID 17401150.
- 1 2 3 4 5 6 Musemeci L, Arthur JW, Cheung FS, Hoque S, Lippman S, Reichardt JK, et al. (January 2010). "Single Nucleotide Differences (SNDs) in the dbSNP Database May Lead to Errors in Genotyping and Haplotyping Studies". Human Mutation. 31 (1): 67–73. doi:10.1002/humu.21137. PMC 2797835. PMID 19877174.
- 1 2 Mitchell AA, Zwick ME, Chakravarti A, Cutler DJ, et al. (2004). "Discrepancies in dbSNP confirmation rates and allele frequency distributions from varying genotyping error rates and patterns". Bioinformatics. 20 (7): 1022–1032. doi:10.1093/bioinformatics/bth034. PMID 14764571.
- 1 2 Carlson CS, Eberle MA, Rieder MJ, Smith JD, Kruglyak L, Nickerson DA, et al. (2003). "Additional SNPs and linkage-disequilibrium analyses are necessary for whole-genome association studies in humans". Nature Genetics. 33 (4): 518–521. doi:10.1038/ng1128. PMID 12652300.
- 1 2 Cutler DJ, Zwick ME, Carrasquillo MM, Yohn CT, Tobin KP, Kashuk C, Matthews DJ, Shah NA, Elchler EE, Warrington JA, Chakravarti A, et al. (2001). "High-Throughput Variation Detection and Genotyping Using Microarrays". Genome Research. 11 (11): 1913–1925. doi:10.1101/gr.197201. PMC 311146. PMID 11691856.
- 1 2 Gabriel SB; Schaffner SF; Nguyen H; Moore J.M; Roy J; Blumenstiel B; Higgins J; DeFelice M; Lochner A; Faggart M; Liu-Cordero SN; Rotimi C; Adeyemo A; Cooper R; Ward R; Lander ES; Daly MJ; Altshuler D; et al. (2003). "The structure of haplotype blocks in the human genome". Science. 296 (5576): 2225–2229. doi:10.1126/science.1069424. PMID 12029063.
- 1 2 Reich DE, Gabriel SB, Altshuler D, et al. (2003). "Quality and completeness of SNP databases". Nature Genetics. 33 (4): 457–458. doi:10.1038/ng1133. PMID 12652301.
- ↑ Dvornyk V, Long JR, Xiong DH, Liu PY, Zhao LJ, Shen H, Zhang YY, Liu YJ, Rocha-Sancher S, Xiao P, Recker RR, Deng HW, et al. (2004). "Current limitations of SNP data from the public domain for studies of complex disorders: a test for ten candidate genes for obesity and osteoporosis". BMC Genetics. 5: 4. doi:10.1186/1471-2156-5-4. PMC 395827. PMID 15113403.
- ↑ de Bakker PI; Yelensky R; Pe’er I; Gabriel SB; Daly MJ; Altshuler D; et al. (2005). "Efficiency and power in genetic association studies". Nature Genetics. 37 (11): 1217–1223. doi:10.1038/ng1669. PMID 16244653.