Buchner ring expansion
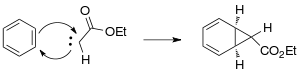
The Buchner Ring Expansion is a two-step organic C-C bond forming reaction used to access 7-membered rings. The first step involves formation of a carbene from ethyl diazoacetate, which cyclopropanates an aromatic ring. The ring expansion occurs in the second step, with an electrocyclic reaction opening the cyclopropane ring to form the 7-membered ring.

History
The Buchner ring expansion reaction was first used in 1885 by E. Buchner and T. Curtius [1][2] who prepared a carbene from ethyl diazoacetate for addition to benzene using both thermal and photochemical pathways in the synthesis of cycloheptatriene derivatives. The resulting product was a mixture of four isomeric carboxylic acids. Variations in the reaction arise from methods of carbene preparation. Advances in organometallic chemistry have resulted in increased selectivity of cycloheptatriene derivatives. In the 1980s it was found that dirhodium catalysts provide single cyclopropane isomers in high yields.[3] Applications are found in medicine (drug syntheses)[4][5][6][7][8] and material science (fullerene derivatives).[9][10][11]
Preparation
Preparation of ethyl-diazoacetate:
Buchner's first synthesis of cycloheptatriene derivatives in 1885 used photolysis and thermal conditions to generate the carbene. A procedure for preparation of the hazardous starting material needed for carbene generation in the Buchner reaction, ethyl-diazoacetate, is available in Organic Synthesis.[12] In the procedure provided, Searle includes cautionary instructions due to the highly explosive nature of diazoacetic esters.
Preparation of the metal carbenoid:
Synthesis of the carbene in the 1960s was focused on using copper catalysts for stereoselective propanation.[13] In the 1980s, dirhodium catalysts have been used to generate the carbenoid for cyclopropanation. The advent of metallochemistry has improved the selectivity of the product ratios of the cyclohexatriene derivatives through choice of ligand on the carbenoid catalyst.[14]
Mechanism
Step 1:
The reaction mechanism of a Buchner ring expansion begins with carbene formation from ethyl-diazoacetate generated initially through photochemical or thermal reactions with extrusion of nitrogen.

The generated carbene adds to one of the double bonds of benzene to form the cyclopropane ring.
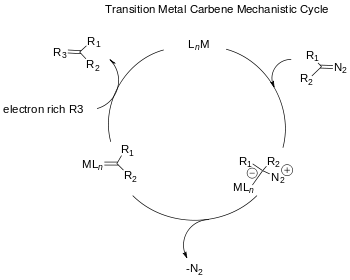
The advent of transition metal catalyzed reagents provides alternative stereospecific methods for cyclopropanation. The choices for metals include Cu, Rh and Ru with a variety of ligands.[13] The use of rhodium catalysts in the Buchner reaction for carbene generation reduces the number of products by producing predominantly the kinetic cycloheptatrienyl esters.[14] Product mixtures of Buchner reactions resulting from thermal Rhodium II-catalysts are less complicated. Wyatt et al. have studied the regioselectivity of the thermal Buchner reaction using Rh2(O2CCF3)4 and demonstrated that the electrophilic character of the rhodium carbene prefers reaction at the more nucleophilic π-bonds of the aromatic ring.[15]

The accepted carbene catalytic cycle[16] was proposed by Yates[17] in 1952. Initially the diazo compound oxidatively adds to the metal ligand complex. Following the extrusion of nitrogen the metal carbene is generated and reacts with an electron rich aromatic substance to reductively regenerate the metal catalyst completing the catalytic cycle.
Step 2:
The second step of the Buchner reaction involves a pericyclic concerted ring expansion. Based on Woodward–Hoffmann rules, the electrocyclic opening of norcaradiene derivatives is a 6-electron disrotatory (π 4s + σ 2s), thermally allowed process.

The norcaradiene-cycloheptatriene equilibrium has been studied extensively.[18] The position of the equilibrium depends upon steric, electronic and conformational effects. Due to conformational strain in the cyclopropane ring of the norcaradiene the equilibrium lies on the side of the cycloheptatriene. The equilibrium may be shifted toward the norcaradiene by destabilization of the cycloheptatriene by bulky substitution (large sterically hindered groups i. e. t-butyl) at C1 and C6.

Equilibrium may be altered by varying substitution at C7. Electron donating groups (EDG) favor the norcaradiene, while electron withdrawing groups (EWG) favor the cycloheptatriene.
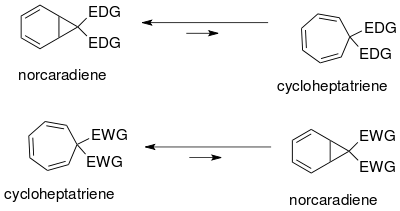
The tautomerism of the norcaradiene and cycloheptatriene can be understood based on the Walsh cyclopropane molecular orbitals of the norcaradiene cyclopropane ring. Electronic rationalization for stabilization of the Walsh orbitals[18] is possible for both electron withdrawing and electron donating groups at the C7 carbon. The molecular orbitals of electron withdrawing groups at C7 overlap with the HOMO Walsh orbitals of the cyclopropane ring causing a shortening of the C1-C6 bond. In the case of electron donating groups, orbital overlap is again possible now in the LUMO, resulting in an increase in antibonding character destabilizing the norcaradiene tautomer. The position of the equilibrium may be controlled depending on the carbene substituents.

Applications
Medicine:
The importance of the Buchner ring expansion annulation chemistry is evident in the application of this synthetic sequence in the synthesis of biological compounds.
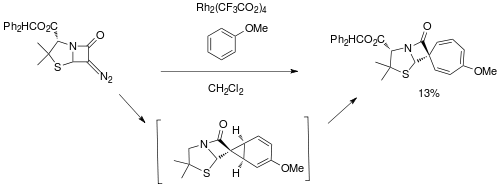
While studying an analogous reaction of carbene addition to thiophene, Stephen Matlin and Lam Chan applied the Buchner ring expansion method in 1981 to generate spiro derivatives of Penicillin.[7]
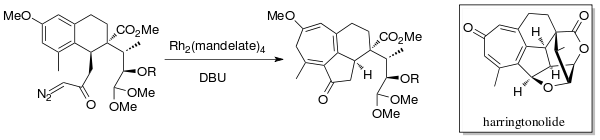
In 1998, Mander et al. synthesized the diterpenoid tropone, Harringtonolide[6] using the Buchner intramolecular ring expansion annulation chemistry. A rhodium catalyst (Rh2(mandelate)4) and DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) were used to generate the carbene. This natural product was found to have antineoplastic and antiviral properties.
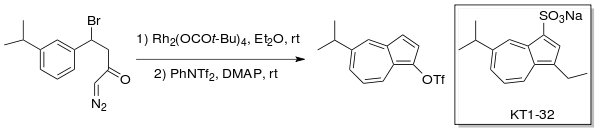
Danheiser et al. utilized intramolecular carbenoid generation to produce substituted azulenes through a Buchner type ring expansion. The anti-ulcer drug, Egualen (KT1-32)[4][5] was synthesized using this ring expansion-annulation strategy with a rhodium catalyst (Rh2(OCOt-Bu)4) in ether.
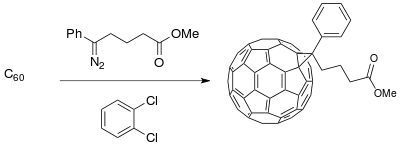
Material Science:
The Buchner ring expansion method has been used to synthesize starting materials for applications in material science involving photovoltaic cells. The development of a donor-acceptor (D-A) interface composed of conducting polymer donors and buckminsterfullerene derivative acceptors create a phase-separated composite that enhances photoconductivity (available with only polymer donors) in the photoinduced charge transfer process of photovoltaic cells.[19] The fullerene compounds can be functionalized for miscibility of C60 to increase efficiency of the solar cell depending upon the polymeric thin film synthesized.[11]
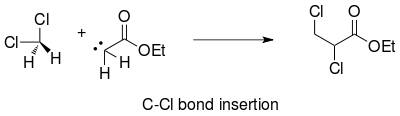
Limitations
The disadvantages of the reaction involve side reactions of the carbene moiety. The choice of solvent for the reaction needs to be considered. In addition to the potential for carbon-hydrogen bond insertion reactions, carbon-halogen carbene insertion is possible when dichloromethane is used as the solvent.[20]
Control for regioselectivity during the carbene addition is necessary to avoid side products resulting from conjugated cycloheptatriene isomers. Noels et al. used Rh(II) catalysts for carbene generation under mild reaction conditions (room temperature) to obtain regioselectively the kinetic non-conjugated cycloheptatriene isomer.[3][8][21]

See also
References
- ↑ Buchner, E.; Curtius, T. (1885), "Ueber die Einwirkung von Diazoessigather auf aromatisch Kohlenwasserstoffe", Ber. Dtsch. Chem. Ges., 18: 2377–2379, doi:10.1002/cber.188501802119
- ↑ Buchner, E.; Curtius, T. (1885), "Synthesis of beta-Keto esters from aldehydes and diazoacetic acid.", Ber. Dtsch. Chem. Ges., 18: 2371–2377, doi:10.1002/cber.188501802118
- 1 2 Hubert, A. J.; Noels, A. F.; Anciaux, A. J.; Warin, R.; Teyssie, P. (1981), "Transition-Metal-CatalyzedReactions of Diazo Compounds. 2.' Addition to Aromatic Molecules: Catalysis of Buchner's Synthesis of Cycloheptatrienes", J. Org. Chem., 46: 873–876, doi:10.1021/jo00318a010
- 1 2 Danheiser, R. L.; J. L. Kane, J.; Shea, K. M.; Crombie, A. L. (2001), "A Ring Expansion−Annulation Strategy for the Synthesis of Substituted Azulenes. Preparation and Suzuki Coupling Reactions of 1-Azulenyl Triflates", Org. Lett., 3: 1081–1084, doi:10.1021/ol0156897
- 1 2 Danheiser, R. L.; Crombie, A. L.; Kane, J. L. J.; Shea, K. M. (2004), "Ring Expansion-Annulation Strategy for the Synthesis of Substituted Azulenes and Oligoazulenes. 2. Synthesis of Azulenyl Halides, Sulfonates, and Azulenylmetal Compounds and Their Application in Transition-Metal- Mediated Coupling Reactions", J. Org. Chem., 69: 8652–8667, doi:10.1021/jo048698c
- 1 2 Mander, L. N.; Frey, B.; Wells, A. P.; Rogers, D. H. (1998), "Synthesis of the Unusual Diterpenoid Tropones Hainanolidol and Harringtonolide", J. Am. Chem. Soc., 120: 1914–1915, doi:10.1021/ja9738081
- 1 2 Matlin, S. A.; Chan, L. (1981), "NEW SPIRO DERIVATIVES OF PENICILLIN", Tetrahedron Letters, 22: 4025–4028, doi:10.1016/S0040-4039(01)82055-4
- 1 2 Reisman, S. E.; Nani, R. R.; Levin, S. (2011), "Buchner and Beyond: Arene Cyclopropanation as Applied to Natural Product Total Synthesis", SynLett, 7: 2437–2442, doi:10.1055/s-0031-1289520
- ↑ Prato, M. (1997), "[60]Fullerene chemistry for materials science applications", J. Mater. Chem., 7: 1097–1109, doi:10.1039/A700080D
- ↑ Wudl, F.; Gonzalez, R.; Hummelen, J. C. (1995), "The Specific Acid-Catalyzedand Photochemical Isomerization of a Robust Fulleroid to a Methanofullerene", J. Org. Chem., 60: 2618–2620, doi:10.1021/jo00113a049
- 1 2 Wudl, F.; Hummelen, J. C.; Knight, B. W.; LePeq, F. (1995), "Preparation and Characterization of Fulleroid and Methanofullerene Derivatives", J. Org. Chem., 60: 532–538, doi:10.1021/jo00108a012
- ↑ Searle, N. E. (1963), "ETHYL DIAZOACETATE", Organic Syntheses, Coll., 4: 424
- 1 2 Lebel, H.; Marcoux, J.; Molinaro, C.; Charette, A. (2003), "Stereoselective Cyclopropanation Reactions", Chem. Rev., 103: 977–1050, doi:10.1021/cr010007e, PMID 12683775
- 1 2 McKervey, A.; Ye, T. (1994), "Organic Synthesis with alpha-Diazocarbonyl Compounds", Chem. Rev., 94: 1091–1160, doi:10.1021/cr00028a010
- ↑ Wyatt, E. E.; Galloway W. R. J. D. & Spring, D. R. (2011), "Regioselectivity in Thermal Rhodium(II)-Catalysed Buchner-Type Reactions of Substituted Aryl Halides: Studies towards the Synthesis of Halide-Substituted Cycloheptatrienes", Synlett, 2011: 1449–1453, doi:10.1055/s-0030-1260562
- ↑ Pirrung, M. C.; Liu, H.; Morehead, J.; Andrew T. (2002), "Rhodium Chemzymes: Michaelis-Menten Kinetics in Dirhodium(II) Carboxylate-Catalyzed Carbenoid Reactions", J. Am. Chem. Soc., 124: 1014–1023, doi:10.1021/ja011599l
- ↑ Yates, P. (1952), "The Copper-catalyzed Decomposition of Diazoketones", J. Am. Chem. Soc., 74: 5376–5381, doi:10.1021/ja01141a047
- 1 2 Maguire, A. R.; McNamara, O. A. (2011), "The norcaradieneecycloheptatriene equilibrium", Tetrahedron, 67: 9–40, doi:10.1016/j.tet.2010.10.030
- ↑ Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J. (1995), "Polymer Photovoltaic Cells: Enhanced Efficiencies via a Network of Internal Donor-Acceptor Heterojunctions", Science, 270: 1789–1791, Bibcode:1995Sci...270.1789Y, doi:10.1126/science.270.5243.1789
- ↑ Lovely, C. J.; Browning, R. G.; Badarinaray, V.; Rasika Dias, H. V. (2005), "A silver-catalyzed Buchner reaction", Tetrahedron Letters, 46: 2453–2455, doi:10.1016/j.tetlet.2005.02.052
- ↑ Doering, W. v. E.; Laber, G.; Vonderwahl, R.; Chamberlain, N. F.; Williams, R. B. (1956), "The structure of the Buchner Acids", J. Am. Chem. Soc., 78: 5448, doi:10.1021/ja01601a080